水溶液及び混合溶媒中でのフェロシアンイオンとオクタシアノモリブデン(V) : イオン間の電子移行に対する陽イオンの効果
10
0
0
全文
(2) (jg 2 SPA) m^- Bgffl62^10^. Journal of Hokkaido University of Education (Section II A) Vol. 38, No. 1 October, 1987. 7]<.WWM^W^(D 7^a->7y^^yfc^-^^>/. ^';yry (v) ^^(DII^WTKW^-^^®^. ^ ffl ^ ff • ^ ^ BS ^ ^w^±w] i is-Kiwm "KjiO^a'^gMb'^. Effects of Cations on the Electron-transfer Reactions between Fe(CN)e4- and Mo(CN)s3- in Water and in Water-organic. Mixed Solvents Sadayuki MATSUDA and Akihiko YAMAGISHI* Chemistry Laboratory, Asahikawa College, Hokkaido University of Education, Asahikawa 070. "'Department of Chemistry, Faculty of Science, Tokyo University, Tokyo 113. Abstract We used a stopped-flow spectrophotometer to study the effects of a cation M+ [=Li+, Na+, K+, Cs+ and N(CH3)+4] on the electron-transfer reaction in water Fe(CN)i,'1- + Mo(CN)83- k. Fe(CN)63- + Mo(CN)s4-. Addition of the electrolyte, M+ C1-, increased the bimolecular rate constant,, k, and the effects were of the order Li+ < Na+ < K+ < N(CNa)4+ < Cs+. The. specific effects of different cations are interpreted in terms of the difference in the degree of salvation of the cations that are involved in a transition state. Because of the dependence of k on the concentration of a cation, we attributed the enhancement of the rate constant to the. contribution of the reaction intermediates. The same reaction was also studied in four kinds of binary solution: water/methanol, water/ethanol, water/acetonitrile and water/dioxane. The composition was so determined that each had an equal dielectric constant of 75. Because of the difference in the K+ ion effects in the investigated solvents, we suggest that the value of k in the mixed solvents is influenced by a selective salvation effect.. (1).
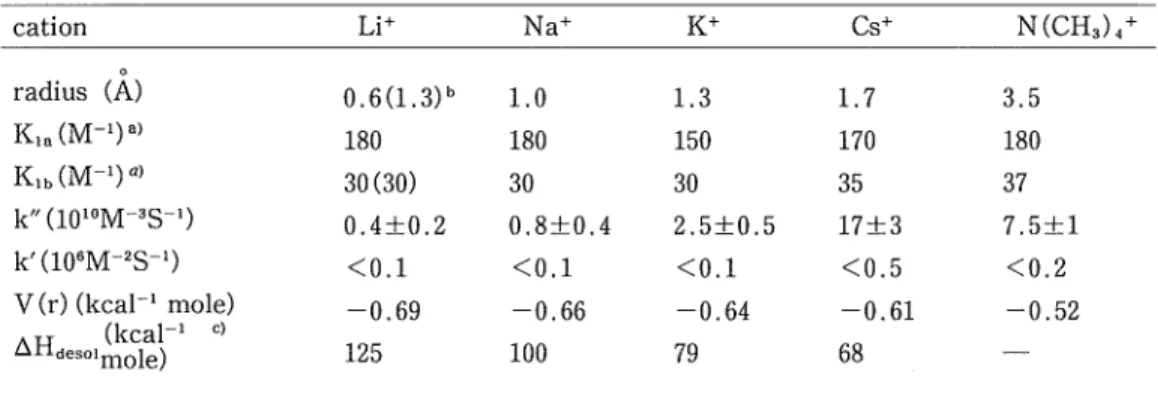
(3) Sadayuki MATSUDA • Akihiko YAMAGISHI. Introduction Several authors1"3' have noted that the rate of the electron exchange between negatively. charged metal complexes like Fe(CN),,4- + Fe(CN)(,3-^ Fe(CN)e3- + Fe(CN)g4- and Mn042 2Mn04-^ Mn04- + Mo042~ is remarkably enhanced by the presence of cations. Since the large cations, which are less solvated, are more effective, it has been suggested that the transition state contains a partly desolvated cation between two exchanging anions and that. this facilitates the formation of less solvated cations"". [1] [Mn04---K+---Mn04]2-. We have recently studied the electron-transfer reaction between Fe(CN)s4- and 7, 7, 8,. 8-tetracyanoquinodimethane or p-chloranil with to test the applicability of the above postulate to the reactions involving organic compounds6'.No evidence that the reaction proceeds via an analogous transition state was found, however. [2] [Fe(CN)6---K+---TCNQ]3-. The observed enhancement of the reaction rate on the addition of K+ was interpreted in terms of the formation of the ion-paired species, KFe(CN)e3- and KFe(CN)g2-.. The present investigation is concerned with the validity of the postulate [1] for the electron exchange reactions between negatively charged metal complexes. In order to examine the cation effects, we have performed a number of kinetic studies of the electron-transfer reaction. [3], Fe(CN)64- + Mo(CN)83-^ Fe(CN)e3- + Mo(CN)84-, in the presence of various kinds of cations.. The same reaction has also been studied in binary solvents of water and organic com-. pounds in order to test whether the observed cation effect can be rationalized in terms of ion-pairing equilibria of reactants or whether it is imperative to assume the participation of a cation in a transition state as in [1].. Experimental K.iMo(CN)8 was prepared according to the formula of Furman and Millers6'. The aqueous. solution of Mo(CN)s3- was obtained by electrolytically oxidizing the solution of K4Mo(CN)g. K4Fe(CN)e was purified by recrystalization from the aqueous solution. KC1 and NaCl was re-crystalized from hydrochloric acid. LiCl, CsCl and N(CHs)4 Cl of G. R. grade were used without further purification. Water was deionized and distlled. Ethanol, methanol, acetonitrile and dioxane were distilled. The binary solvents with the dielectric constant of 75 were prepared according to the equation:.
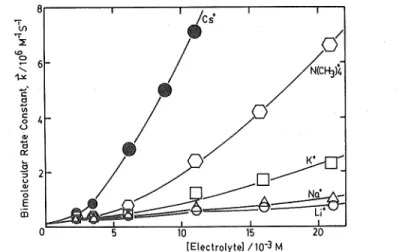
(4) electron-transfer between Fe(CN)6-1- and Mo(CN)83- 3. [4] 75=DiWi + D^Wz where Di and Dz are the dielectric constants of water and an organic compound, respectively, and Wi and Wz the respective weight percentage.. The reaction rate of [3] was measured with a stopped-flow spectrophotometer, Union Giken RA-13007). The progress of the reaction was monitored by the increase of absorbance. at 420 nm (Ama.v of Fe(CN)6"l-). The association constants of K+ with Fe(CN)63- and with ]V[o(CN)s3- were measured by potentiometric titration. The activity of K+ ion was measured with an Orion K+ sensitive electrode, model 92-19. All the measurements were performed at 20°C.. Results and Discussion (a) Cation Effects on Fe(CN),,-1- -Mo(CN)s3- in Aqueous Solution. Because of the standard oxidation-reduction potential of the couple Fe(CN)64-/Fe(CN)( 6 3and Mo(CN)84-/Mo(CN)83~ 8>, the equilibrium constant of [3] is estimated to be 5X106 at room temperature. Thus, when two solutions of Fe(CN)e'4- and Mo(CN)a3- of the same concentra-. tion are mixed, the reaction passes over to the righthand side completely. The forward rate of reaction [3] was measured under the condition of the initial concentration of reactants [K< Fe(CN)e]o = [Ka Mo(CN)Jo = 2.96 XlO-4 M. The bimolecular rate constant of [3] is calculated by the equation,. [5] k=(t^[X]o)-1 2. in which tj_ is the half-life time of reaction and [X]o the initial concentration*'. In Fig. 2. 1, the k thus obtained is shown at various concentrations of added electrolytes. Anions are known to have little effects on the reactions between negatively charged complexes2'. The. observed increase of k on the addition of the electrolytes can therefore be attributed to the effect of cations. Since the reacting complexes are potassium salts, the background concentration of K+ is 2.0 X 10~3 M. The presence of K+ seems not to obscure the effect of the added cations. For all the cases k increases super-linearly with the increase of M+. For the. quantitative analysis of the kinetic data, we need to know the degree of association of the cations with the present reactants. The association constants Kia and Kib of the following. equilibria were therefore estimated from the potential change during the titration by the 1.0 M solution of KC1.A typical potential change is shown in Fig. 2. [6a] Fe(CNV- + K+^ KFe(CN)63- ; Kia *' The actual initiation is estimated to occur about 1 msec after "mixing" with the present apparatus.. (3).
(5) Sadayuki MATSUDA • Akihiko YAMAGISHI. 20. 10—i5(Electrolyte)/10-3M. Fig. I. The dependence of the bimolecular rate constant, k, on the concentration of various electrolytes : (0) LiCl; (A) NaCl;. (D) KC1; (0) N(CH3)4C1 ; and (•) CsCl.. [6b] Mo(CN)83- + K+^ KMo(CN)a. Ki. -30. Applying an ordinary procedure9', we obtain from the arrest at Log [K+]/M = -2.6 Kia =. 150 ± 30M-' and Kii, = 30 ± 10M-' for [6b]. According to the Bjerrum theory of ion-pairing, the values of Kia and Kib are estimated respec-. tively to be ca. 40 and 15, if the attractive force is electrostatic and the closest distance is assumed to be 8 A and 9A, respectively. These values are of the same order of the experimental ones, which suggests that the K+—associations. -2.8 -2.6. with Fe(CN)e4- and with Mo(CN)83- occur mainly as a result of electrostatic force. The experimental values of Kia and Kib mean that appre-. ciable fractions of both the reactants are present as ion-paired species under the conditions of. Log [K*1/M. Fig. 2.. The typical plot of the potential. change of the K+ ion selective electrade referred to a calomel electrode against the logarithm of added K+ ion. [K<Fe(CN)e]=2.50xlO-4 M.. kinetic measurements. We therefore assume. that the following paths are involved in the present electron-transfer reaction [3].. [ 7 ] Fe(CN)e + Mo(CN)83MFe(CN)o3- + Mo(CN)g3[8a] (4).
(6) electron-transfer between Fe(CN)g4- and Mo(CN)s. [ 8 b ] Fe(CN)o4- + MMo(CN)8i!- k,b. [ 9 ] MFe(CN)e3- + MMo(CN)s2- k^ Then, the overall rate constant k is represented as. [10] k =. ko + k'[M]+ k"[M]2 1+ K'[M]+ K"[M]2. where [M] is the formal concentration of cation, and k', k", K' and K" are related to the. rate constants of [8a]-[9], and the association constants of [6] as below: k' = kia Kia + kib Klb k" = k. K,a Kib K' = Kia + Ki, K" - K,a Kib. We know all the parameters in eq. [10] except kia, kib and ks, if M+ = K+, since Kia and. Kib are both known, ko is obtained at [M] = 0 in Fig. 1. The quantity [k(l + K' [K+] + K" [K+]2)—ko]/[K+]is thus plotted against [K+] as shown in Fig. 3. The figure shows that each plot falls roughly on a straight line, whose intercept and slope give k' and k", respectively. Similar analyses were also made for other cations, where K' and K" are estimated from. the theoretical values of Kia and Kib (see the captions of Table 1). The ko, k' and k" thus obtained are compiled in Table 1. The very small value of k' implies that the reaction proceeds almost exclusively through the paths of [7] and [9] under the present conditions. Since Kig and Kib do not vary by more than two steps for the added cations, k" can be. directly related to kz, the rate constant of reaction path [9]. As we can see in Fig. 1, the effect of cations are in the order L|+ < Na+ < K+ < N(CH3).i+ < Cs+. This sequence agrees almost. 10 15 tK<l/10-3M. Fig. 3. Plot of the quantity, [k(l+ K'[K+] + K"[K+]2) -k»]/[K+ against [K+] according to eq. [10].. (5).
(7) Sadayuki MATSUDA • Akihiko YAMAGISHI Table I. k" for various kinds of cations. Li+. cation. K+. Na+. Cs+. N(CH3)<+. radius (A). 0.6(1.3)". 1.0. 1.3. 1.7. 3.5. K,,(M-')a). 180. 180. 150. 170. 180. K,,(M-l)a>. 30(30). 30. 30. 35. 37. k" (1010M-3S->). 0.4±0.2. 0.8±0.4. 2.5±0.5. 17±3. 7.5±1. k'(10"M-2S-1). V(r)(kcal-1 mole) (kcal-' c> ldeso'mole). <0.1. <0.1. <0.1. <0.5. <0.2. -0.69. -0.66. -0.64. -0.61. -0.52. 125. 100. 79. 68. a) Estimated from the Bjerrum theory of Ki=C.47rNa3/(3xlO'). exp (-ZiZ,e2/DkTa), where a is the sum of the radii of reactant ion and cation. The correction factor C is added so as to give the agreement between the theoretical and experimental values in case of K+ ; C^S and 2 for Km and Kib, respectively.. b) The radius of solvated Li+ ion. c ) The enthalpy change of desolvation of a cation for the reaction, M+(aqueous)<±M+ (gaseous), taken from Ref. 4, p. 81.. exactly with that of their ionic radius. The fact that kg varies with the ionic radius is interpreted as follows. This order will have been the result of the following two opposing effects. The first is the attractive effect of the cation in the formation of the transition state, when one of the ion-paired cations is supposed to intervene between two negative reactants so. as to reduce the electrostatic repulsion. This stabilizing effect can be roughly estimated if we assume the following three-particle system ;. [11]. The potential energy, V(r) is given by - 3. - 3. (r> = [~S+7 ^ "VTF + 17+2r> ~e. where the radii of MFe(CN)63- and Mo(CN)g3- are taken as 8 and 9 A, respectively and e is the dielectric constant of the enviroment of reactants. The value of V(r) thus caculated is given for each cation in Table 1. The smaller the cation is, and the more negative the V(r), the more effective it will be in stabilizing the intermediate structure [11]. The other effect is the difference in the salvation energy of the cation. When a cation participates in the formation of the activated state [II], it will be desolvated, partially at least. As we can evidently see from the enthalpy of the salvation of a cation which is given in the lowest rank of Table 1, the smaller the radius of a cation is, the more energy will be required to desolvate the cation. In conclusion, it must be noted that the intermediate structure includes the ion-paired species, MFe(CN)<,3- or MMo(CN)s2-.. (6 ).
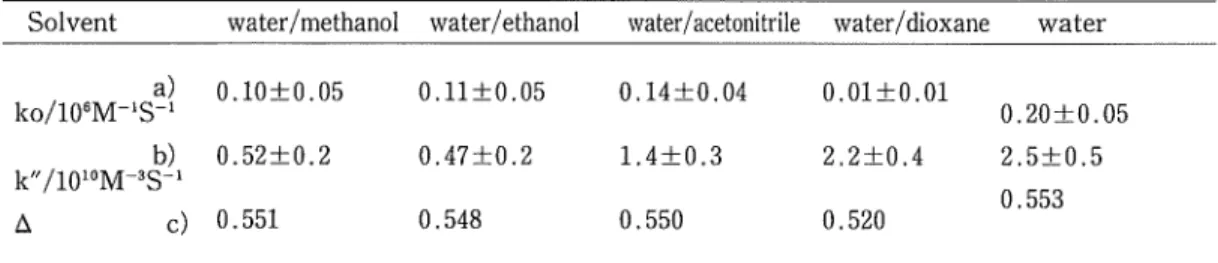
(8) electron-transfer between Fe(CN)e4- and Mo(CN)ss. (b) K+ Effects on Fe(CN)e4- -Mo(CN)83- in Binary Solutions We also studied the reaction [3] in four kind of binary solution: water/methanol, water/ethanol, water/acetonitrile and water/ dioxane ; at the same time the effect of K+ on the rate constant was examined. All the mixtures were adjusted to have the same dielectric constant, 75, as that mentioned in the experimental section. In Fig, 4, the bimolecular rate constant, k, similarly. obtained, is plotted against the concentration of KC1. In these experiments, KC1 adds up. 4 6 [KC11/10-3M. to 8 X 10~3 M. Since, in this range of concentration, the added electrolyte dissociates5',. [KC1] in Fig. 4, is equal to that of [K+] . The. Fig. 4.. enhancement of the rate constant is seen for. The dependence on the concentration of K+ of the bimolecular rate constant, k, in the binary solvents : (A) water/ethanol;. (0) water/mothanol ; (•) water/. all solvents with the increase of added K+ ion.. acetonitrile ; and (Q) water/dioxane.. This increase of k can again be ascribed to the involvement of ion-paired species in the electron-transfer (scheme [7] — [9]). By assuming the negligible contribution of paths [8a] and [8b] to the overall rate, the quantity/[k(l+ K' [K+] + K"[K+]2)-ko]/[K+] is plotted against [K+] ; this was done in pure water. Here, Kia and Kib in the binary solvents are estimated with the Bjerrum electrostatic theory, and ko is obtained by extrapolating k to [K+]:=0. From the slope of an obtained straight line, k" is determined as given in Table 2. We note that both ko and k" depend considerably on the nature of the solvents. Two other points are noteworthy, (i) ko in a binary solvent is smaller than in water; ko in a water/. Table 2. ko and k" for various kinds of solvents Solvent. water/methanol water/ethanol water/acetonitrile water/dioxane water. A. 0 .14±0.04. 0 .01±0 .01. 0.52±0. 2. 0 .47±0 .2. 1 .4±0.3. 2 .2±0. 4. 0.551. 0 .548. 0 .550. 0 .520. 0.10+0.. b). k"/10 10M-3S-1. c). 05. 0 ,n±o .05. a!. ko/10 8M-'S-1. 0 .20±0.05 2 .5±0.5 0 .553. a ) The values of k obtained by extrapolation to [K+]:=0. b) The slope of the plot of [k(l+K'[K+]+K"[K+]2)-k,]/ [K+] against [K+] , where Ki. and K,,, are estimated to be 250M-'and 40M~1, respectively, from the Bjerrum electrostatic theory. C ) A=WI (Dop'.B-Ds^) +Wz (Dop',^i-D,;U, where Wi and W; are the weight percentages of water and organic solvent, respectively, and. D,p and D, are the optical and static dielectric constants, respectively. Suffixes w and org denote water and organic solvent, respectively.. 7).
(9) 8 Sadayuki MATSUDA • Akihiko YAMAGISHI. dioxane medium is especially small. (ii) k" in a binary solvent is smaller than it is in water, although a water/dioxane medium gives a relatively high value of k". It is well-known that the ionic association phenomenon of binary solvents such as those. used in the present study have been described in terms of the "sphere in dielectric continuum" model"'. That is to say, the electrostatic interactions, i.e. the repulsion between negative ions or the formation of ion-paired species, are determined solely by the value of s. On the other. hand, according to the Marcus' electron-transfer theory which is based equally on the dielectric continuum model, the activation free energy increases linearly with A = Dop-l — Ds-l, where Dop and Ds are optical and static dielectric constants, respectively 12). The last rank of Table. 1 gives the value of A ; this was calculated on the assumption that each component of the binary solvents contributes to A independently in proportion to its weight percentage. It can be seen that the A valve of water and binary solvents is similar, except for the relatively small value of A for water/dioxane. Both ko and k" can therefore be expected to be higher in the water/ dioxane mixture than those of other solvents. This however, was not observed for the ko values.. The above argument lead us to the conclusion that the present kinetic results can not be interpreted within the framework of the conventional dielectric continuum theory. In other words, one has to take into account the selective salvation of reacting ions and K+ with either the water or the organic components.. Thus, if dioxane molecules dominate in solvating the reacting ions, the local dielectric constant in the vicinity of the reactants is lower than 75 ; i.e. the electrostatic repulsion between Fe(CN)64- Mo(CN)83- is higher, and hence the ko value is smaller than that expected from the dielectric continuum theory. The higher value of k" in water/dioxane is similarly interpreted. In the reaction path [9], the K+ ion acts as a bridge between the two negatively charged reactants. If the K+ ion is partially desovated in its activated state [II], because the desolvation energy of K+ is lower in the organic solvents than in water13', we shoud expect that the organic molecules are desolvated. Accordingly, the formation of the activated state becomes. easier. The large value of ka in the water/dioxane medium is therefore consistent with the fact that the dielectric constant of dioxane is the lowest and hence the poorest solvating medium for K+ ion.. References 1) J. C. Sheppard and A. C. Wahl, /. Amer. Chem. Soc., 79, 1020 (1957). 2 ) M. Shporer, G. Ron, A. Lowenstein and G. Navon, Inorg. Chem., 4, 361 (1965). 3 ) R. J. Campion, C. F. Deck, P. King and A. C. Wahl, Inorg. Chem., 6, 672 (1967). 4 ) F. Basolo and R. G. Pearson, "Mechanirn of Inorganic Reactions". Willey, New York. (1967), p. 477. 5 ) S. Matsuda and A. Yamagishi, Can. ]. Chem., 56, 2216 (1978)..
(10) electron-transfer between Fe(CN)s<- and Mo(CN)s3- 9. 6 ) N. H. Furman and C. 0. Miller, Inorg. Syn., 3, 160 (1950).. 7 ) A. Yamagishi, Bull. Chem. Soc. Japan, 47, 2152 (1974). 8) W. M. Latimer, "Oxidation Potentials" 2nd ed, Prentire-Hall, Englewood Cliffs, N. J.,. (1964). 9 ) e.g. E. S. Amis, "Solvent Effects on Reaction Rates and Mechanism", Academic Press,. New York (1966), p. 273. 10) N. Bjerrum, Kgt. danske Vidensk, Selsk. 4, 26 (1906). 11) H. K. Bodensen and J. B. Ramsey, /. Rhys. Chem., 69, 543 (1965). 12) R. A. Marcus, /. Chem. Phys., 24, 966 (1956). 13) D. A. Owensky, A. J. Parker and J. W. Diggle, /. Amer. Chem. Soc., 95, 2682 (1973).. 9).
(11)
図
![Fig. 3. Plot of the quantity, [k(l+ K'[K+] + K"[K+]2) -k»]/[K+](https://thumb-ap.123doks.com/thumbv2/123deta/6736767.1156909/6.774.153.582.719.948/fig-plot-quantity-k-k-k-k-k.webp)
関連したドキュメント
Significant variations in total arsenic concentrations in different fractions of raw rice (hull, 271 .. endosperm, polished rice, whole rice, and bran) have been reported
aripiprazole水和物粒子が徐々に溶解するのにとも ない、血液中へと放出される。PP
If, as argued above, monetary transfers between the water utility and potential customers disconnected are not allowed, then the water utility will be required to satisfy
Kyoto University Research Information Repository https://repository.kulib.kyoto-u.ac.jp... A Self-archived
4 because evolutionary algorithms work with a population of solutions, various optimal solutions can be obtained, or many solutions can be obtained with values close to the
These authors successfully used the llnearlzed theory for calculation of wave loading on a vertical circular cylinder extending from a horizontal ocean floor to above the free
Zhang; Blow-up of solutions to the periodic modified Camassa-Holm equation with varying linear dispersion, Discrete Contin. Wang; Blow-up of solutions to the periodic
A large amount of friction and heat transfer data, for different values of the dimensionless pitch and height with square, rectangular, trapezoidal and triangular shape ribs, has
