研究で
“分子モデリング” を
初めて導入するコツ
分子モデリング導入セミナー 第1回
2008年3月6日
於 和光純薬工業株式会社 東京支店
Habata Research Group Department of Chemistry
& Research Center for Materials
with Integrated Properties Toho University
Japan
幅田 揚一
化学
物質の学問
経験によって分子を設計
合成
物性評価
新しい分子の創生/従来の方法
新しい分子
3新しい分子
新しい分子の創生/計算化学
経験によって分子を設計
理論計算
性質を予測
合成
物性評価
4波動関数による計算方法の確立 電子密度による計算方法の確立5
1998年度
ノーベル化学賞
分子モデリングとは,コンピュータを使って以下のような 情報を得る手法の総称 1. 孤立した分子や分子集合体についての定量的な情報 2. 分子間の相互作用や反応についての定性的な情報 3. 反応機構の説明に使うことができる遷移状態の構造 4. 平衡状態のエネルギーの差から得られる反応のエネルギー (熱化学).熱化学的な計算.とくに結合の生成,開裂を伴う 反応の全エネルギーや絶対的活性化エネルギー 5. 平衡状態のエネルギーと遷移状態のエネルギーの差から得 られる活性化エネルギー(動力学)
分子モデリングで何がわかるか
W. J. Hehre, J. Schnitker, A. J. Shusterman, Molecular Modeling in Undergraduate Chemistry
1. 原子間距離,角度,体積などを即座に計測できる 2. 最も低いエネルギーを持つ分子の構造を推定できる 3. 最も高いエネルギーを持つ分子(遷移状態や不安定な分子) を推定できる 4. 分子軌道法,電荷,静電ポテンシャルによって反応しやすい場 所を推定できる 5. 様々な分子の性質(物理的,化学的性質)を予測することがで きる 6. 等値面,プロパティーマップなどのグラフィックスやアニメーショ ンを利用して情報の視覚化ができる
分子モデリングで(具体的)に何ができるか
7分子モデリングのための
基礎知識
非経験的分子軌道法(HF) 完全に量子力学計算を行う.経験的なパラメータは使用しな い 密度汎関数法(DFT) 量子力学的なエネルギーを電子密度の分布を用いて計算す る 半経験的分子軌道法(Semi empirical) 経験的なパラメータを使用して量子力学計算を行う 分子力場(MM) 経験的なパラメータを使用して物理学的な計算を行う
理論モデルの種類
量子力学計算とはひたすらSchrödinger波動方程式を解くこと 9理論モデルをどうやって選ぶか
理論モデルの選択(1) 分子力場モデル 1. 平衡構造計算 2. コンホメーション解析 半経験的分子軌道モデル 1. HFやDFTで行うと時間がかかりすぎるような巨大分子 の平衡構造や遷移状態構造の計算 2. DFTで行うと時間がかかりすぎるような遷移金属を含 む無機・有機化合物.HFでは遷移金属を含む化合物 に対しては良好な結果を与えない 3. 反応エネルギー計算と配座エネルギー計算は不可 10W. J. Hehre, J. Yu, P. E. Klunzinger, L. Lou, A Brief Guide to Molecular Mechanics and
理論モデルの選択(2) Hartree-Fockモデル 1. DFT計算では時間がかかりすぎるような中程度の大き さの有機分子や典型元素を含む無機分子の構造決定 半経験的分子軌道計算よりも好ましい結果を与える 2. 反応エネルギー計算 半経験的分子軌道計算では好ま しい結果を与えない 3. 絶対的な 活性化エネルギー,結合の生成や開裂を伴 う反応,遷移金属を含む無機・有機分子の計算は不可
理論モデルをどうやって選ぶか
11W. J. Hehre, J. Yu, P. E. Klunzinger, L. Lou, A Brief Guide to Molecular Mechanics and
理論モデルの選択(3) DFTモデル 1. HFモデルでは計算できない大きな分子や半経験的分 子軌道計算では精密さが不十分な分子 2. HFモデルでは精密さが不十分な分子 3. 熱化学的な計算.とくに結合の生成,開裂を伴う反応 や絶対的活性化エネルギー計算 4. とても小さな系や非常に平坦なポテンシャルエネルギー 局面を持つ系の平衡構造や遷移構造計算は不可
理論モデルをどうやって選ぶか
12W. J. Hehre, J. Yu, P. E. Klunzinger, L. Lou, A Brief Guide to Molecular Mechanics and
DFT 力場 半経験的 HF SVWN pBP 構造最適化 (有機分子) GC G G G G 構造最適化 (遷移金属) P G G G P 遷移状態構造 N/A G G G G コンホメーション G P GC G GC 熱化学 (非isodesmic) N/A P GC G GC 熱化学 (isodesmic) N/A P GC G GC 時間コスト 極めて低い 低い 中程度 中程度 高い 各モデルのパフォーマンスと時間コスト
G = good GC = good with cautious application P = poor N/A = not applicable
力場 半経験的 DFT HF SVWN pBP 構造最適化 (有機分子) GC G G G G 構造最適化 (遷移金属) P G G G P 遷移状態構造 N/A G G G G コンホメーション G P GC G GC 熱化学 (非isodesmic) N/A P GC G GC 熱化学 (isodesmic) N/A P GC G GC 時間コスト 極めて低い 低い 中程度 中程度 高い 力場 半経験的 DFT HF SVWN pBP 構造最適化 (有機分子) GC G G G G 構造最適化 (遷移金属) P G G G P 遷移状態構造 N/A G G G G コンホメーション G P GC G GC 熱化学 (非isodesmic) N/A P GC G GC 熱化学 (isodesmic) N/A P GC G GC 時間コスト 極めて低い 低い 中程度 中程度 高い 力場 半経験的 DFT HF SVWN pBP 構造最適化 (有機分子) GC G G G G 構造最適化 (遷移金属) P G G G P 遷移状態構造 N/A G G G G コンホメーション G P GC G GC 熱化学 (非isodesmic) N/A P GC G GC 熱化学 (isodesmic) N/A P GC G GC 時間コスト 極めて低い 低い 中程度 中程度 高い
理論モデルをどうやって選ぶか
W. J. Hehre, A. J. Shusterman, W. W. Huang, A Laboratory Book of Computational Organic
Chemistry, Wavefunction, Inc. (2002)
計算にかかる時間 計算内容 エネルギー 構造最適化 振動数 equatrialメチルシクロヘキサン 7重原子,Cs対称,32独立変数 AM1 0.01 0.08 0.66 3-21G 1 14 190 6-31G* 5.4 90 1100 リセルグ酸 20重原子,C1対称,102独立変数 AM1 0.05 1.9 11 3-21G 17 600 -6-31G* 120 -
-理論モデルをどうやって選ぶか
14W. J. Hehre, A. J. Shusterman, W. W. Huang, A Laboratory Book of Computational Organic
• 多次元のエネルギー面上の局所的な 部位における最小値を探すことです • 原理的には,どんなエネルギー最小 値の構造(これを平衡構造といいます) でも実験によって得ることができます. 計算によって得られた構造の“質”を 判定することができます
平衡構造
15量子化学計算と比較して非常に簡単なため、分子力場法は配座解 析に早くから使われてきました.分子力場は最安定コンホマーを求 めたりコンホマー間のエネルギー差を求めることには適しません。 数多くの配座の中から好ましい構造を選択することに適しています
配座解析
Cyclohexane 反応で得られる異性体の安定性の比較 A+B → α+β のとき,異性体間のエネルギー差(安定性の差) ΔEISOMER は ΔEISOMER = Eα- Eβ 16• 反応経路上のエネルギー最 大値の場所とその構造を探 索することです • 遷移状態の構造は実験で は検出不可能であり,計算 でのみ得ることができます
遷移状態構造
・遷移状態であることの確認 ① 振動計算で虚の振動が1個ある こと(通常800-2000 cm-1程度) ② 虚の振動の基準振動が生 成物と反応物をスムーズにつなぐ動きであること 17• 遷移状態を探し、そのエネルギー の二次微分から振動を得ることが できます • 振動の動きは、遷移構造を中心と した反応座標系になっています。 • この動き(反応物と生成物をなめら かに結ぶことが必要)を実験的に 再現することは不可能であり,遷 移構造を探索する手法は他には ありません
反応経路
19• 平衡構造におけるエネルギーの二次微分はIRの振動を導きます • この振動の動きは鉛筆でも表現できます • 複雑な分子の場合、コンピュータ分子モデリングだけが振動の動き を表現することができます 20
振動数
2 12
1
⎥
⎦
⎤
⎢
⎣
⎡
+
=
My
Mx
MxMy
f
c
π
ν
ν
=波数(cm-1) c=光速度(cm s-1) f=結合の力の定数(dyn cm-1) Mx,
My=原子Mx,Myの質量(g)• ここでの目的は二つのエネル ギー最小値の値の差を求める ことです • 「通常の」反応熱の計測(例え ば燃焼)を元にした生成エンタ ルピーから得ることができます • 計算値の検証が可能です
反応エネルギー
反応のエネルギーの計算方法 A+B+C+ ・・・ → α+β+γ+ ・・・ のとき,反応の全エネルギー ΔEREACTION は次のように計算さ れる ΔEREACTION = (Eα+Eβ+Eγ+ ・・・) – (Ea+Eb+Ec + ・・・) 21• 熱力学支配による生成物は反応経路にかかわらずエネルギー が最小になる生成物であり、速度論支配による生成物はその 生成物のエネルギーにかかわらず遷移状態のエネルギーが最 小になる反応経路を経て生成される化合物です • この系は熱力学支配や速度論支配の意味や反応の遷移状態 の扱いに対して実際的な経験を得るのために助けとなります またこのような経験を積むことで 反応機構に習熟することができ るようになります
反応の熱力学的支配と速度論的支配
22• 活性化エネルギーの差によっ て速度論的な生成物の生成 比がわかります • 全エネルギーの差によって熱 力学的な生成物の生成比が わかります 相対的な活性化エネルギーの計算(速度論支配) 可逆反応の場合 α ← Tα‡ ← A+B→ T β‡ → β のとき,
活性化エネルギー差ΔETRANSITION STATE= ETβ‡– E Tα‡
速度論的異性体の生成比Nα/Nβ = exp -1060( ETα‡- ETβ‡ ) 相対的なエネルギーの計算(熱力学支配) 可逆反応の場合 X ←A+B→ Y のとき, 熱力学的異性体の生成比Nx/Ny = exp -1060( E - E ) 23
速度論的な生成物と熱力学的生成物
• 電子の存在確立の分布(電 子密度)を計算することがで きます • 電子密度等値面はその値 を選ぶことにより,原子の位 置、結合、分子の大きさな どの様々な情報を提供して くれます • 分子の大きさと形を表現し ます.同様の情報はCPKモ デルでも示すことができま す
電子密度
24電子密度
Cyclohexanone 0.1 electrons/au3 isodensity Cyclohexanone 0.002 electrons/au3 isodensity Diborane 0.115 electrons/au3 isodensity 25• 静電ポテンシャル⇒分子近傍に正の点電荷(求電子試薬)を おいたときのポテンシャルエネルギー • 静電ポテンシャルの負値の等値面⇒求電子攻撃を受けやす い場所 • ベンゼンにおける負値の静電ポテンシャル面は、ベンゼンの 面の上および下からのp軌道において求電子反応の可能性を 示しています • ピリジンの場合は環のp軌道ではなく、窒素原子のs軌道を求 電子試薬が攻撃することを示しています。 • 静電ポテンシャルを見ることで構造の似ている二つの分子の 振る舞いが大きく異なることが分かります
静電ポテンシャル
26静電ポテンシャル
Benzene Pyridine
• 静電ポテンシャル面から、その領域が反応し易いかどうかを 判断することは易しくありません • 電子密度面上に静電ポテンシャルを色分けしてマップを作成 すると分かりやすい表示方法となります • 作成されたモデルは、分子の大きさ、形と同時に、分子表面 上の静電ポテンシャルを提示できます • 色分けは、最小値(負の値)を赤で、最大値(正の値)を青で 表示しています • ベンゼンのモデルではπ電子系、ピリジンではσ電子系を示 唆する部位が赤く色分けがされており、この部分で求電子反 応が起きると説明することができます
静電ポテンシャルマップ
28静電ポテンシャルマップ
Ethanol Acetic acid Nitric acid Benzene
単位
1. HF,DFT計算のエネルギー: au 2. 半経験的分子軌道計算のエネルギー: kcal/mol 3. 分子力場計算によるひずみエネルギー: kcal/mol 4. 軌道エネルギー: eV 5. 電子密度とスピン密度: electrons/au3 6. 双極子モーメント: debyes 7. 原子の電荷: electrons 8. 静電ポテンシャル: kcal/mol 9. 局所イオン化ポテンシャル: eV 10. 振動数: wavenumbers (cm-1) 重要な単位換算1au = 627.5 kcal/mol = 2625 kJ/mol (1 cal ≒ 4.184 J)
分子モデリングの
利用例
1) 分子構造:分子構造の表示 2) 安定構造:1H NMRの解釈と認識場 3) 分子構造:立体障害による反応生成物の説明 4) 静電ポテンシャル:孤立電子対の張り出し 5) 配座解析と安定構造:X線結晶構造との合致 6) エネルギー計算とNBO解析: Ag+-π相互作用 7) HOMOとLUMO:Ag+-π相互作用の視覚化(非公開) 8) 静電ポテンシャル: 1H NMRの解釈と錯体の構造 9)静電ポテンシャル: ベンゼン環の電子密度 10)静電ポテンシャルマップ:ベンゼン環の電子密度(非公開) 11)ジアステレオマーの構造:生成比の見積(非公開)
分モデリングの利用例
32分モデリングの利用例1 (分子構造)
O O O O O P O HO O J. Org. Chem., 1994, 59, 676. Chem3D 33分子モデリングの利用例 2 (安定構造)
J. Org. Chem., 1996, 61, 8391.
CAChe
分子モデリングの利用例 3 (立体障害)
O O Cl Cl O O O O O HO O O O O O O OCH3 O O O H3CO HO O O OCH3 O O O O O O O Hd O O O O O O OCH3 O Hc Hb Ha Hd He He O O O Cl O O OCH3 HO O O O O O O O O O O O O O Cl O O Hc Hb Ha Hd He Hf S NaH / THF 1 (36 %) 5 S S S NaH / THF 2 (19 %) + + 6 + 1:1 mixture (total 8 %) 2.4 equiv. 4 (63 %) 2.4 equiv. 1.2 equiv. S NaH / THF 1 (11 %) 3 (44 %) S S + S 1.5 equiv. NaH / THF A mixture of 2 (12 %) and 3 (43 %) 2.3 equiv. NaH THF Recover (76 %) of 1,8-dichloroanthraquinone Chem3D 3-21G Tetrahedron., 1997, 53, 4179. 35O O N O O N OH 1: n = 0 2: n = 1 O O HN O O (i) HCHO/MeOH (ii) benzene N n OH n N O H N O (a) (b) N N O O J. Heterocyclic Chem., 2001, 38, 253. 静電ポテンシャル等値面
分子モデリングの利用例 4 (静電ポテンシャル)
36分子モデリングの利用例 5 (配座解析と安定構造)
N O O O O OH HO N O O OH HO N O O O O OH HO 1 2 3J. Chem. Soc., Perkin Trans. 1, 2001, 38, 253.
Result of conformational search
The podands that form pseudo-cyclic structures by intramolecular hydrogen bonding
Podand 2
Optimized structure
using 3-21G(*)
Crystal structure
The podands that form pseudo-cyclic structures by intramolecular hydrogen bonding
Podand 2
Crystal structure
Optimized structure using 3-21G(*)
Podand 3
The podands that form pseudo-cyclic structures by intramolecular hydrogen bonding
分子モデリングの利用例 6 (エネルギーとNBO解析)
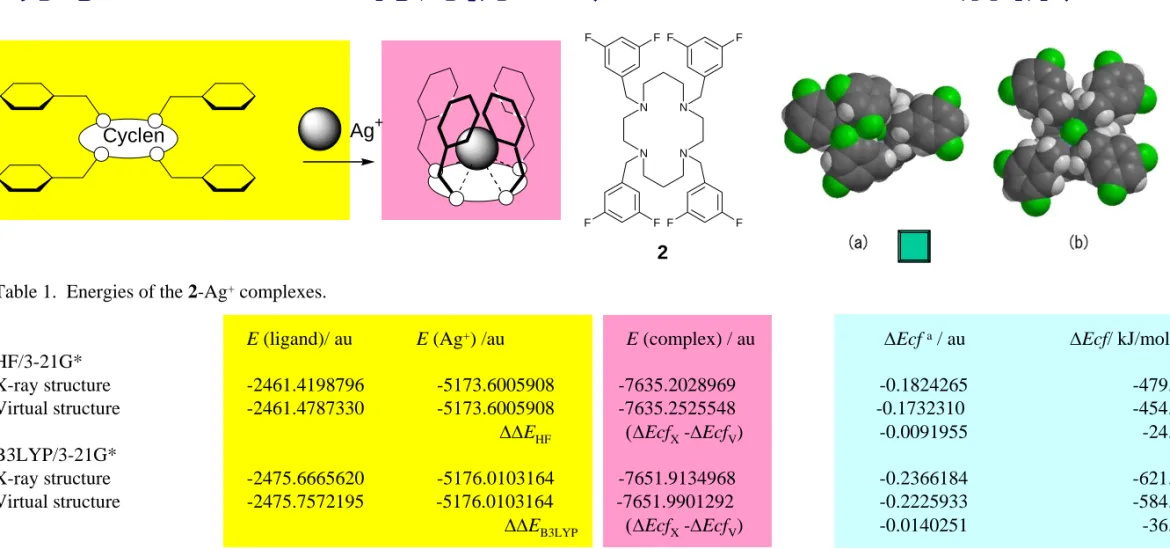
Ag+ Cyclen
Table 2. Results of the NBO analysis of Ag+in the X-ray and virtual structures of the 2-Ag+complex at the HF/3-21G(*) theoretical level.
4d orbitals (10) 5s orbital (0) net change X-ray structure 9.9461 (-0.0539) 0.1133 (+0.1133) +0.0594 Virtual structure 9.9480 (-0.0520) 0.1073 (+0.1073) +0.0553
J. Inclusion Phenom. Macrocycl. Chem., 2004, 49, 17.
N N N N F F F F F F F F 2
Table 1. Energies of the 2-Ag+complexes.
E (ligand)/ au E (Ag+) /au E (complex) / au ΔEcfa/ au ΔEcf/ kJ/mol b
HF/3-21G*
X-ray structure -2461.4198796 -5173.6005908 -7635.2028969 -0.1824265 -479.0 Virtual structure -2461.4787330 -5173.6005908 -7635.2525548 -0.1732310 -454.8 ΔΔEHF (ΔEcfX-ΔEcfV) -0.0091955 -24.2
B3LYP/3-21G*
X-ray structure -2475.6665620 -5176.0103164 -7651.9134968 -0.2366184 -621.2 Virtual structure -2475.7572195 -5176.0103164 -7651.9901292 -0.2225933 -584.4 ΔΔEB3LYP (ΔEcfX-ΔEcfV) -0.0140251 -36.8
aΔEcf = E (complex) – E (ligand) – E (Ag+). ΔEcf
XandΔEcfVareΔEcf for X-ray structure and virtual structure, respectively. b1 au =2625.5 kJ/mol
分子モデリングの利用例 8 (静電ポテンシャル)
O S N S 3a 4a S S N S N N O S N S 3b 4b S S N S N N3a-AgOTf 4a-AgOTf 3b-AgOTf 4b-AgOTf
Dalton Trans., 2006, 1836.
分子モデリングの利用例 9 (静電ポテンシャル)
S N S S S N S S OCH3 S N S S CH3 S N S S S N S S S N S S F F Cl Cl NO2 2a 2b 2c 2d 2e 2f Inorg. Chem., 2007, 46, 6529. 43分子モデルリング
利用にあたって
分子モデリングは “絵に描いた餅である” “とても時間がかかる” “できるものとできないものがある” “発散することが多い” “真空中に1個だけ存在しているときの挙動である” “初期構造が大事” などやっかいな点が多いですが “説得力がある説明ができる” “論文にワンセンテンス加えることができる” ので是非利用してみてください 45