大規模並列量子化学計算プログラム
SMASHの開発と応用計算
石村 和也
(分子科学研究所 ポスト「京」重点課題5)
[email protected]
[email protected]
第7回材料系ワークショップ
〜第一原理計算と実験のデータ同化による材料開発手法〜
2019年2月8日
本日の内容
• 量子化学計算の概要
• 分子科学分野スパコンの変遷
• SMASHプログラムについて
• SMASHプログラムの性能
• 応用計算
• まとめ
量子化学計算とは
• 量子化学計算:
分子系の電子分布やエネルギーを計算
し、
分子の構造、反応性、物性などを解析・予測する
• 入力:原子の電子分布 (原子軌道)、原子座標
• 出力:分子の電子分布 (分子軌道)
• 計算量は計算方法により異なり、原子数の3乗から7乗(もしく
はそれ以上)に比例して増加する
原子軌道 (C,H)
原子座標
Hartree-Fock計算
H
H
H
H
H
H
分子軌道 (ベンゼン(C
6H
6))
量子化学計算で得られるもの
• 分子のエネルギー
• 安定構造、遷移状態構造
• 化学反応エネルギー
• 光吸収、発光スペクトル
• 振動スペクトル
• NMR(核磁気共鳴)スペクトル
• 各原子の電荷
• 溶媒効果
• 結合軌道解析など
反応前
A+B
反応後
C+D
遷移状態
反応座標
エ
ネル
ギー
活性化エ
ネルギー
生成熱
触媒
化学反応 A+B → C+D
量子化学計算方法とコスト
•
演算内容から分類
計算方法
演算量
データ量
通信量
Hartree-Fock(SCF), DFT法
2電子積分計算(キャッシュ内演算)
密対称行列の対角化
O(N
4)
(カットオフで
O(N
3)程度)
O(N
2)
O(N
2)
摂動(MP2,MP3,..)法,
結合クラスター(CCSD, CCSD(T),..)法
密行列-行列積
O(N
5~)
O(N
4~) O(N
4~)
配置間相互作用(CIS, CISD,...)法
疎行列の対角化
O(N
5~)
O(N
4~) O(N
4~)
原子の数が 2倍になると、計算量はN
3:8倍、
N
5:32倍
原子の数が10倍になると、計算量はN
3:1,000倍、N
5:100,000倍
計算コストを減らすため、分割法や近似の導入が数多く提案されている
N:原子(or基底)数
分子科学分野スパコンの変遷1
•
自然科学研究機構 岡崎共通研究施設 計算科学研究センター (旧分子科学研究所
電子計算機センター)におけるCPU能力の変遷
https://ccportal.ims.ac.jp/
年
理論総演算性
能 (TFLOPS)
1979
3.6x10
-51989
0.002
1999
0.092
2009
13
2015
492
2017
4,076
演算性能は10年で数百倍のペースで向上
分子科学分野スパコンの変遷2
•
自然科学研究機構 岡崎共通研究施設 計算科学研究センター (旧分子科学研究所
電子計算機センター)におけるCPU能力の変遷
https://ccportal.ims.ac.jp/
年
機種
理論総演算性能
(TFLOPS)
1979 HITACHI M-180 (2台)
3.6 x 10
-52000 IBM SP2 (48 台)
0.01
NEC SX-5 (8CPU)
0.06
Fujitsu VPP5000 (30PE)
0.28
SGI SGI2800 (256CPU)
0.15
合計
0.51
2017 NEC LX (Xeon 20コア x 2, 2.4GHz) (
32560 コア
)
2,500
NEC LX (Xeon 18コア x 2, 3.0GHz) (
5724 コア
)
549
NEC LX (Xeon 12コア x 2, 3.0GHz) (
1536 コア
)
221
(+
NVIDIA Tesla P100 x 2
)
806
合計
4,076
地球シミュレータ(2002)
35
京コンピュータ(2011)
10,510
←Core i7 7700K
(4.2GHz,4コア)
2台分
← 京コンピュータ
の約半分
✓ 並列計算は必要不可欠で、大規模並列計算も当たり前の時代になりつつある
✓ 2017年10月には、演算性能で京コンピュータ並のマシンが稼働
SMASHプログラム
•
大規模並列量子化学計算プログラムSMASH (Scalable Molecular Analysis
Solver for High performance computing systems)
•
オープンソースライセンス
(Apache 2.0)
•
http://smash-qc.sourceforge.net/
•
2014年9月1日公開
•
対象マシン:スカラ型CPUを搭載した計算機
(PCクラスタから京コンピュータまで)
•
エネルギー微分、構造最適化計算を重点的に整備
•
現時点で、Hartree-Fock, DFT(B3LYP), MP2計算が可能
•
MPI/OpenMPハイブリッド並列を設計段階から考慮したアルゴリズム及びプ
ログラム開発 (Module変数、サブルーチン引数の仕分け)
•
言語はFortran90/95
•
1,2電子積分など頻繁に使う計算ルーチンのライブラリ化で開発コスト削減
•
電子相関計算の大容量データをディスクではなくメモリ上に分散保存
•
2014年9月から2019年1月までの約4年半で、ダウンロード数は約1500
SMASH ウェブサイト
最新ソースコードと
日本語マニュアル
SMASH
プログラム開発にあたって
分子サイズ
計算精度
DFT
MP2
CCSD
CCSD(T)
Gaussian
GAMESS
Molpro
Turbomole
NWChem
…
SMASHの現時点でのカバー領域
計算・開発両面で使いやすくするために
✓ シンプル(インプット、実行方法、ライセンス)
✓ トラブル(収束しない、インプットが間違っている)で計算をストップす
る場合、エラーメッセージと対処方法を出力
HF
SMASHに取り入れたこれまでの取り組み
高速化
D
C
B
A
x
y
z
P
Q
(AB|CD)
K. Ishimura, S. Nagase, Theoret Chem Acc, 2008, 120, 185.
漸化式を用いて効率的に高い軌道角
運動量の2電子積分を計算する方法
と座標軸回転により演算量を減らす
方法の組み合わせ
GAMESSのデフォルトルーチンとして
使われている
並列化
K. Ishimura, K. Kuramoto, Y. Ikuta, S. Hyodo, J. Chem. Theory Comp. 2010, 6, 1075.
!$OMP parallel do schedule(dynamic,1) do m=n, 1, -1 <--- OpenMPによる振り分け
do n=1, m
mn=m*(m+1)/2+n
lstart=mod(mn+mpi_rank,nproc)+1
do l=lstart, m ,nproc <- MPIランクに
よる振り分け do s=1, l AO2電子積分(mn|ls)計算+ Fock行列に足し込み enddo enddo enddo enddo call mpi_allreduce(Fock)
MPI/OpenMP2段階並列化
原子軌道Gauss関数
Hartree-Fock計算Hartree-Fock法
11(
) (
)
−
+
=
s l l s mn mnmn
ls
ml
ns
, ,|
|
2
i i iC
C
H
F
原子軌道(AO)2電子積分
εSC
FC =
Fock行列
F: Fock行列, C: 分子軌道係数
S: 基底重なり行列,
e: 分子軌道エネルギー
初期軌道係数C計算
AO2電子反発積分計算+
Fock行列への足し込み (O(N
4))
Fock行列対角化 (O(N
3))
計算終了
分子軌道C収束
分子軌道
収束せず
𝜇𝜈|𝜆𝜎 = න 𝑑𝒓
1න 𝑑𝒓
2𝜙
𝜇𝒓
1𝜙
𝜈𝒓
11
𝑟
12𝜙
𝜆𝒓
2𝜙
𝜎𝒓
2𝜙
𝜇𝒓
1: 原子軌道Gauss関数
原子軌道の線形結合係数
(分子軌道係数)を求める
12
MPI/OpenMP並列アルゴリズム
!$OMP parallel do schedule(dynamic,1) reduction(+:Fock)
do
m=nbasis, 1, -1
<-- OpenMPによる振り分け
do
n=1, m
mn=m(m+1)/2+n
lstart=mod(mn+mpi_rank,nproc)+1
do
l=lstart, m ,nproc
<-- MPIランクによる振り分け
do
s=1, l
AO2電子積分(
mn|ls)計算+Fock行列に足し込み
enddo
enddo
enddo
enddo
!$OMP end parallel do
call mpi_allreduce(Fock)
Fock行列
=
+
(
) (
−
)
s l l s mn mnmn
ls
ml
ns
, ,|
|
2
i i iC
C
H
F
原子軌道(AO)2電子積分
K. Ishimura, K. Kuramoto, Y. Ikuta, S. Hyodo, J. Chem. Theory Comp. 2010, 6, 1075.
do
m=1, nbasis
do
n=1, m
do
l=1, m
do
s=1, l
AO2電子積分(
mn|ls)計算
+Fock行列に足し込み
enddo
enddo
enddo
enddo
並列化前
MPI/OpenMPハイブリッド並列化後
✓ MPI
と
OpenMP
でFock行列計算を並列化
B3LYP(DFT)エネルギー並列計算性能
0
10000
20000
30000
40000
50000
60000
0
24576
49152
73728
98304
CPUコア数
Sp
eed
-up
CPUコア数
6144
12288
24576
49152
98304
実行時間 (秒)
1267.4
674.5
377.0
224.6
154.2
✓ 10万コアで5万倍のスピードアップ、実行性能13%
✓ 10万コアで360原子系のB3LYP計算が2分半
✓ 行列対角化(LAPACK,分割統治法)3回分の時間は約35秒
→ScaLAPACK、EigenExaなどプロセス並列化されているライブラリ導入が今後必要
計算機 : 京コンピュータ
分子
: (C
150H
30)
2(360原子)
基底関数 : cc-pVDZ
(4500基底)
計算方法 : B3LYP
SCFサイクル数 : 16
Hartree-Fockエネルギー1ノード計算性能
基底関数
GAMESS
SMASH
6-31G(d) (1032 基底)
706.4
666.6
cc-pVDZ (1185 基底)
2279.9
1434.3
GAMESSとの比較
• Xeon E5649 2.53GHz 12core、1ノード利用
• Taxol(C
47H
51NO
14)のHartree-Fock計算時間(sec)
• 同じ計算条件(積分Cutoffなど)
✓ 1ノードでは、GAMESSよりHartree-Fock計算時間を最大40%削減
✓ SMASHではS関数とP関数を別々に計算するため、SP型の基底では
B3LYP(DFT)エネルギー1次微分並列計算性能
CPUコア数
1024
4096
8192
16384
微分計算のみの
実行時間(秒)
402.0
101.2
50.8
25.5
計算機 : 京コンピュータ
分子
: (C
150H
30)
基底関数 : cc-pVDZ (2250 functions)
計算方法 : B3LYP
✓ エネルギー計算と同様にエネルギー1次微分計算についても
MPI/OpenMP並列アルゴリズムを開発
✓ エネルギー計算と異なり対角化計算が無いため、並列化効率は
ほぼ100%
0.0
4096.0
8192.0
12288.0
16384.0
0
4096 8192 12288 16384
CPUコア数
Spee
d-up
2次の摂動(MP2)法1
✓ Hartree-Fock計算で分子のエネルギーの約99%を求
めることができるが、定量的な議論を行うためには
残り1%の電子相関エネルギーが重要
✓ MP2法は最も簡便な電子相関計算方法
✓ 積分変換(密行列-行列積)計算が中心
𝑎𝑖|𝑏𝑗 =
𝜇𝜈𝜆𝜎 𝐴𝑂𝐶
𝜇𝑎𝐶
𝜈𝑖𝐶
𝜆𝑏𝐶
𝜎𝑗(𝜇𝜈|𝜆𝜎)
𝐸
𝑀𝑃2=
𝑖𝑗 𝑜𝑐𝑐
𝑎𝑏 𝑣𝑖𝑟𝑎𝑖|𝑏𝑗 2 𝑎𝑖|𝑏𝑗 − 𝑎𝑗|𝑏𝑖
𝜀
𝑖+ 𝜀
𝑗− 𝜀
𝑎− 𝜀
𝑏(
mn
|
ls
)計算 (O(N
4))
(
m
i|
ls
)計算 (O(N
5))
(
m
i|
l
j)計算 (O(N
5))
(ai|
l
j)計算 (O(N
5))
(ai|bj)計算 (O(N
5))
MP2エネルギー計算 (O(N
4))
Hartree-Fock計算
𝜀
𝑖: 軌道エネルギー, 𝐶
𝜇𝑎: 分子軌道係数
原子軌道(AO)
2電子積分
分子軌道(MO)
2電子積分
2次の摂動(MP2)法2
✓ MP2法は、Hartree-Fock法で記述できない非共有結合(分散力、π-π相
互作用など)を取り扱える
MPI/OpenMP並列アルゴリズム
✓ MPI
と
OpenMP
でMP2計算を並列化
✓ 中間データをメモリに蓄えて、ディスクは不使用
✓ 任意のメモリ量で計算実行可能(ただし、メモリ使用量を削減すると演算量
が増える場合がある)
do ij-block
do
ml (AO index pair)
!$OMP parallel do
do s
AO積分計算
(mn|ls) (all n)
第1変換
(mi|ls) (partial i)
enddo
!$OMP end parallel do
第2変換(dgemm)
(mi|lj) (partial j)
end do ml
do partial-ij (MO index pair)
MPI_sendrecv
(mi|lj)
第3変換(dgemm) (
mi|bj) (all b)
第4変換(dgemm) (ai|bj) (all a)
MP2エネルギー計算
end do ij
end do ij-block
call mpi_reduce(MP2エネルギー)
𝑎𝑖|𝑏𝑗 =
𝜇𝜈𝜆𝜎 𝐴𝑂𝐶
𝜇𝑎𝐶
𝜈𝑖𝐶
𝜆𝑏𝐶
𝜎𝑗(𝜇𝜈|𝜆𝜎)
𝐸
𝑀𝑃2=
𝑖𝑗 𝑜𝑐𝑐
𝑎𝑏 𝑣𝑖𝑟𝑎𝑖|𝑏𝑗 2 𝑎𝑖|𝑏𝑗 − 𝑎𝑗|𝑏𝑖
𝜀
𝑖+ 𝜀
𝑗− 𝜀
𝑎− 𝜀
𝑏MP2エネルギー計算のメモリ使用量と実行時間
✓ 全プロセス合計の使用メモリ量はO
2N
2/2 → O
2N
2/(2
n
ij-block)
(O:占有軌道数、N:基底関数次元数, n
ij-block:占有軌道分割数)
✓ メモリ使用量削減のための追加演算コストは比較的小さい
– 計算条件:Taxol (C
47H
51NO
14)、MP2/6-31G(d) (O=164軌道、 N=970基底)
– 計算機:Fujitsu PRIMEGY CX2500 (Xeon E5-2697, 28コア/ノード) 1ノード
占有軌道分割数
1
2
3
MP2計算時間
765.7
943.4
1121.5
そのうちAO2電子
積分と第1変換
618.8
803.6
981.2
メモリ使用量
101
51
34
計算時間(秒)とメモリ使用量(GB)
20%, 40% up
50%, 67% down
MP2エネルギー1次微分計算性能
✓ エネルギー計算と同様にエネルギー1次微分計算についても
MPI/OpenMP並列アルゴリズムを開発
✓ 使用ノード数が増えるほどトータルのメモリ量は増えて、占有軌道
分割数が減るため、使用ノード数以上の並列加速率が得られた
– 計算条件:C
150H
30、MP2/cc-pVDZ
– 計算機:「京」
MP2エネルギー微分計算時間(秒)と並列加速率
コア数
1536
3072
6144
12288
占有軌道分割数
3
2
1
1
MP2微分実行時間
4253.6
2002.0
743.6
382.0
並列加速率
1536.0
3263.2
8785.6
17103.2
構造最適化回数の削減
Cartesian
座標
Redundant
座標
Luciferin(C
11H
8N
2O
3S
2)
63
11
Taxol (C
47H
51NO
14)
203
40
Table B3LYP/cc-pVDZ構造最適化回数(初期構造HF/STO-3G)
Taxol (C
47H
51NO
14) Luciferin(C
11H
8N
2O
3S
2)
✓ Redundant座標と力場パラメータを使い、初期ヘシアンを改良する
ことでCartesian座標に比べて最適化回数を1/5から1/6へ削減
✓ 2サイクル目以降のヘシアンはBFGS法で更新
SMASHのコンパイル方法
✓ 必要なコンパイラはFortran、ライブラリはBLAS・LAPACK
✓ 並列化はノード間がMPI(MPIライブラリが必要)、ノード内はOpenMP(コン
パイラオプションで指定)
1. ダウンロードしたファイルを展開
tar xfz smash-2.2.0.tgz
2. 展開でできたディレクトリに移動
cd smash
3. makeを実行
ifortベースのmpif90を使う場合:make
mpiifortを使う場合:make –f Makefile.mpiifort
京やFX100を使う場合:make –f Makefile.fujitsu
ifortを使う場合: make –f Makefile.x86_64.noMPI
SMASHの実行方法
✓ MPI(ノード間)並列
✓ OpenMP(ノード内)並列
•
bashの場合:~/.bashrcファイルに次の行を追加して
次のコマンドを実行
•
tcshの場合:~/.tcshrcファイルに次の行を追加して
次のコマンドを実行
export OMP_NUM_THREADS=(スレッド数)
ulimit -s unlimited
export OMP_STACKSIZE=1G
setenv OMP_NUM_THREADS (スレッド数)
unlimit
setenv OMP_STACKSIZE 1G
mpirun -np (プロセス数) ./bin/smash < (inputファイル名) > (outputファイル名)
source ~/.bashrc
SMASHのインプット
✓ サンプルインプット・アウトプットファイルはsmash/example/を参照
✓ 計算方法や条件をjob, control, scf, opt, dft, mp2セクションで設定
✓ 分子座標はgeom行の次から記入、空行もしくはファイルの最後で座標読み込み終了
✓ 基底関数やECPを元素ごとに指定する場合、basis行、ecp行の次から記入
✓ 原子核の電荷(点電荷)の指定はcharge行の次から記入
✓ 大文字と小文字の区別は無し
job runtype=optimize method=b3lyp basis=6-31g(d)
geom
O 0.0000000 0.0000000 0.1423813
H 0.0000000 0.7568189 -0.4626257
H 0.0000000 -0.7568189 -0.4626257
SMASH計算結果の可視化
• 可視化
– Version1では、フリー可視化ソフトParaViewでMOを表示させるためvtk
ファイルを作るプログラムを用意
– Version2からは、GaussViewなどでも表示できるようcubeファイルを作る
プログラムも用意
– 現在、X-Ability社のWinmostarでも対応を進めている
SMASHがインストールされているスパコン
•
インストール済み
– 名大(CX400)
– 九大(ITO-A,ITO-B)
– 東工大(TSUBAME3.0)
– FOCUS
•
インストール中
– 東北大(LX 406Re-2)
– JCAHPC(Oakforest-PACS)
– 東大(Reedbush-U)
– 京大(XC40)
– 阪大(OCTOPUS)
•
詳しくは、次のページを参照
– http://www.hpci-office.jp/pages/appli_software/
– https://www.j-focus.jp/benchmark/
Crystal Structure of Selenolate-Protected Au
24
(SeR)
20
Nanocluster
Yongbo Song, Shuxin Wang, Jun Zhang, Xi Kang, Shuang Chen, Peng Li, Hongting Sheng,
and Manzhou Zhu*
Department of Chemistry, Anhui University, Hefei, Anhui 230601, P. R. China
*
S Supporting InformationABSTRACT: We report the X-ray structure of a selenolate-capped Au24(SeR)20 nanocluster (R = C6H5). ItexhibitsaprolateAu8kernel, which can beviewed astwo
tetrahedral Au4unitscross-joined together without sharing
any Au atoms. The kernel is protected by two trimeric Au3(SeR)4 staple-like motifs as well as two pentameric Au5(SeR)6staplemotifs. Compared to thereported gold− thiolate nanocluster structures, the features of the Au8
kernel and pentameric Au5(SeR)6 staple motif are
unprecedented and provide a structural basis for under-standing the gold−selenolate nanoclusters.
T
hiolate-stabilized gold nanoclusters have attracted wide research interest in recent years. To date, a number of size-discrete gold nanoclusters have been identified,1−14 and a few of them have been structurally characterized by single-crystal X-ray single-crystallography.15−23In parallel with the thiolate-protected gold nanoclusters, recent works have revealed that, bychanging theligand of thegold nanoclustersfromthiolateto selenolate (HSeR), more-stable gold nanoclusters can be produced, and the related properties have been studied.24−27 These studies also found that selenolate-protected Aun(SeR)mnanoclusters possess characteristics different from those of the Aun(SR)mcounterpartsand thushaveconsiderable potential as new functional nanomaterials. However, there have been no reports thus far on the successful crystallization of Aun(SeR)m
nanoclusters. In order to clarify theprecisecorrelation between the ligand and cluster stability, the structure of nanoclusters protected by selenolate should be pursued. Herein we report the first structure of selenolate-stabilized Au24(SeC6H5)20 nanoclusters.
Details of the synthesis are provided in the Supporting Information. Briefly, HAuCl4·3H2O wasdissolved in water and
then phase-transferred to CH2Cl2 with the aid of
tetraocty-lammonium bromide (TOAB). Then, both C6H5SeH and NaBH4 were added simultaneously to convert Au(III) into Au(I) or Au(0) by co-reduction. After reaction overnight, the aqueousphasewasremoved. Themixturein theorganicphase was rotavaporated, and then washed several times with CH3OH/hexane. Dark brown crystals were crystallized from
CH2Cl2/ethanol over 2−3 days. The crystals were then collected. The structure of Au24(SeC6H5)20 was determined by X-ray crystallography. The optical absorption spectrum of Au24(SeC6H5)20nanoclusters(dissolved in tolueneor CH2Cl2)
showsthreestepwisepeaksat380, 530, and 620nm(Figure1). Of note, the optical spectrum of the thiolate counterpart, i.e., Au24(SC2H4Ph)20 nanoclusters (dissolved in toluene or
CH2Cl2), shows a distinct band at 765 nm and a shoulder band at 400 nm.28
The total structure of the Au24(SeC6H5)20 nanocluster is
shown in Figure2. Asimilar structurewasdiscussed in previous
DFT calculations by Pei et al. on thiolate-capped Au24(SR)20 nanoclusters.29 To find out details of the atom-packing structure, we focus on the Au24Se20 framework without the
carbon tails (Figure 3A). The Au24Se20 can be divided into a
prolate Au8 kernel (Figure 3A, highlighted in green), two trimeric Au3Se4 staple-like motifs (Figure 3B, labeled i and
highlighted with blue curves), and two pentameric Au5Se6
staple motifs (Figure 3B, labeled ii and highlighted with blue curves). Following this anatomy, the Au24Se20 framework can
Received: December 25, 2013 Published: February 18, 2014
Figure 1. Optical absorption spectrum of Au24(SeC6H5)20 nano-clusters.
Figure 2. Crystal structure of a selenophenol-protected Au24(SeC6H5)20nanocluster. (Color labels: yellow = Au, violet = Se, gray = C; all H atoms are not shown).
Communication pubs.acs.org/JACS
Se-Protected Au
24
(SePh)
20
Nanocluster
Selenolate-Protected Au Nanocluster; Au
24
(SePh)
20
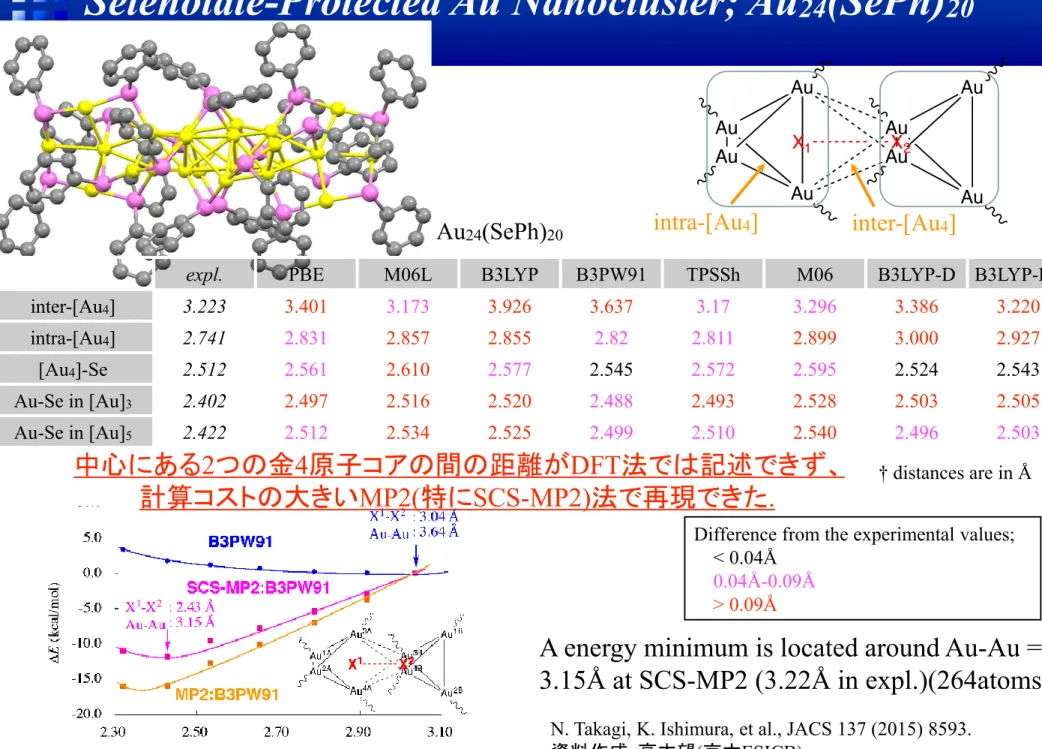
expl. PBE M06L B3LYP B3PW91 TPSSh M06 B3LYP-D B3LYP-D3 inter-[Au4] 3.223 3.401 3.173 3.926 3.637 3.17 3.296 3.386 3.220
intra-[Au4] 2.741 2.831 2.857 2.855 2.82 2.811 2.899 3.000 2.927
[Au4]-Se 2.512 2.561 2.610 2.577 2.545 2.572 2.595 2.524 2.543
Au-Se in [Au]3 2.402 2.497 2.516 2.520 2.488 2.493 2.528 2.503 2.505
Au-Se in [Au]5 2.422 2.512 2.534 2.525 2.499 2.510 2.540 2.496 2.503
Difference from the experimental values; < 0.04Å 0.04Å-0.09Å > 0.09Å
inter-[Au
4]
intra-[Au
4]
† distances are in Å中心にある
2つの金4原子コアの間の距離がDFT法では記述できず、
計算コストの大きい
MP2(特にSCS-MP2)法で再現できた.
Au
24(SePh)
20A energy minimum is located around Au-Au =
3.15Å at SCS-MP2 (3.22Å in expl.)(264atoms).
N. Takagi, K. Ishimura, et al., JACS 137 (2015) 8593. 資料作成:高木望(京大ESICB)
Models of Core-shell, Alloy and Phase-separated
Structures of Cu
32
Ru
6
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
コアシェル型
固溶体合金型
相分離型
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
Ru
N. Takagi, K. Ishimura, et al., JPCC 121 (2017) 10514.
Relative Stability of Cu
32
M
6
M = Ru, Rh, Os, Irの場合、Mをコアとしたコアシェル型構造が安定
(M = Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au)
Cu
32Ru
6 (triplet)Cu
32Rh
6 (singlet)Cu
32Pd
6 (singlet)Cu
32Ag
6 (triplet)Cu
32Os
6 (singlet)Cu
32Ir
6 (triplet)Cu
32Pt
6 (singlet)Cu
32Au
6 (triplet)0.0
0.0
0.0
0.0
0.0
0.0
0.0
0.0
(in kcal/mol)
コアシェル型
固溶体合金型
+16.4
+19.5
-6.1
-16.9
+33.0
+29.4
-5.5
-23.7
+34.8
+18.3
-7.6
-19.0
+76.5
+38.9
-12.4
-26.9
+80.0
+47.0
-33.4
-109.6
+157.3
+93.6
-51.6
-141.2
+101.1
+104.3
+128.9
+139.1
+24.9
-108.1
+35.3
-97.6
相分離型
M
M
M
M
M
Core-shell Structure of Cu
32
M
6
Cu
32Ru
6 (triplet)Cu
32Rh
6 (singlet)Cu
32Pd
6 (singlet)Cu
32Ag
6 (triplet)Cu
32Os
6 (singlet)Cu
32Ir
6 (triplet)Cu
32Pt
6 (singlet)Cu
32Au
6 (triplet)Mのd
電子
ポピュレーション (e)
Cu
32シェルの構造不安定化
エネルギー (kcal/mol)
M-M 距離 (Å)
Cu
38 (triplet)8.06 (
+1.06
)
8.90 (
+0.90
)
9.62 (+0.62)
9.90 (
-0.10
)
7.72 (
+0.72
)
8.66 (
+0.66
)
9.60 (+0.60)
9.88 (
-0.12
)
9.86 (-0.14)
19.9
23.9
40.3
46.8
22.9
22.3
50.9
61.7
0.0
2.684 (+0.112)
2.772 (+0.200)
2.880 (
+0.308
)
2.837 (
+0.265
)
2.657 (+0.085)
2.738 (+0.166)
2.946 (
+0.374
)
2.937 (
+0.365
)
2.572 (0.0)
CTによる安定化
変形による不安定化
コアシェル構造
Ru, Rh, Os, Ir
大きい
小さい
➠
安定
Pd, Ag, Pt, Au
小さい
大きい
➠
不安定
Stable Structure of Cu
32
M
6
Cluster
PIMD-SMASHの開発
✓ PIMDプログラム(志賀)とSMASHプログラム(石村)の組み合わせ
✓ 一体化したプログラムで、使いやすさと高い並列化効率を実現
✓ オープンソースライセンス(Apache 2.0)で公開
S. Ruiz-Barragan, K. Ishimura, M. Shiga, Chem. Phys. Lett. 646 (2016) 130.
水溶液中でDA 反応が加速される:実験 DDG = 4 kcal/mol Breslow et al. J. Amer. Chem. Soc. 102 (1980) 7816.
Diels-Alder反応 (cyclopentadiene + butenone)
Minimum energy path - String method with 120 images. B3LYP/6-31G*