活性化血液凝固第 IX因子は EGFドメインを 介してアポトーシスを誘導する
日本大学大学院医学研究科博士課程 生理系分子細胞生理学専攻
石川 友美
修了年 2016年
指導教員 國分 眞一朗
活性化血液凝固第 IX因子は EGFドメインを 介してアポトーシスを誘導する
日本大学大学院医学研究科博士課程 生理系分子細胞生理学専攻
石川 友美
修了年 2016年
指導教員 國分 眞一朗
目次
概要 1
第一章 緒言
第1節 背景 3 1. アポトーシス
2. EGFモチーフ
3. 凝固第IX因子(coagulation factor IX; F9)
第2節 本研究の目的
第二章 対象と方法 8 第1節 細胞と培養
第2節 試薬
第3節 細胞形態の観察 第4節 免疫組織化学染色 第5節 細胞接着の評価
第6節 Lactate dehydrogenase(LD)活性測定 第7節 アネキシンV染色
第8節 ウェスタンブロット法によるEGF-F9の影響の評価 第9節 カスパーゼ3阻害剤存在下でのLD活性測定
第10節 カスパーゼ3阻害剤存在下でのアネキシンV染色 第11節 活性化カスパーゼ3陽性細胞の評価
第12節 接着阻害培養
第13節 カスパーゼ3阻害剤存在下での細胞接着の評価
第14節 カスパーゼ3阻害剤存在下でのMitogen-activated protein Kinase(MAPK)のタンパク産生の検出
第15節 統計分析
第三章 結果 18 第 1節 細胞接着への影響
第 2節 アポトーシスの誘導
第 3節 情報伝達のカスパーゼ 3依存性 第 4節 MAPKのリン酸化
第四章 考察 22
第1節 EGF-F9の情報伝達路 第2節 EGF-F9の医学的意味 第3節 アミノ酸配列CXDXXXXYXCXC 第五章 まとめ 26
謝辞 27
図 28
図説 37
引用文献 41
研究業績 46
1
本文
概要
目的
凝固第IX因子は血液凝固に必須のタンパクである。その第一上皮成長因子
(epidermal growth factor; EGF)ドメインはCXDXXXXYXCXCのアミノ酸配列を 持ち、このモチーフにはアポトーシス誘導能があると報告されている。このド メインによるアポトーシス誘導能について検討する。
方法
ポリメラーゼ連鎖反応(polymerase chain reaction;PCR)により凝固第IX 因子の第一EGFドメイン(EGF−F9)をコードする配列を増幅しAPtag4ベクターに 挿入した。作成したプラスミッドをChinese Hamster Ovary細胞に遺伝子導入し てアルカリフォスファターゼタグ付きの組み替えタンパクを作成し、実験に用 いた。ヒト重層扁平上皮癌由来の細胞株A431NSの培養液中に100nMのEGF-F9を 添加し、細胞に振盪刺激を加えて細胞基質間接着を評価した。また、免疫染色 やウェスタンブロットを用いてアポトーシス誘導能を評価した。さらに、ウエ スタンブロットを行いEGF-F9により誘導されるアポトーシスにかかわるタンパ クを同定した。同定されたタンパクの阻害剤を使用し、EGF-F9による細胞接着 の減弱化やアポトーシスの誘導がどのように変化するか評価した。
2
結果
EGF−F9を作用させるとA431NS細胞の細胞基質間接着は減弱化し、アポトーシ スが誘導された。この機能は、凝固反応の過程と同様に、F9から活性化ペプチ ドが除去されることにより活性化された。EGF−F9によるカスパーゼ3活性の亢進 は、刺激後5分から10分で起きた。さらに、EGF−F9の投与によりリン酸化p38MAPK が増加していた。アポトーシスはカスパーゼ3阻害剤により部分的に抑制された。
カスパーゼ3阻害剤はEGF-F9による細胞脱接着とリン酸化p38MAPKの増加も抑制 した。
結論
EGF-F9により、細胞の脱接着が起こり、アポトーシスが誘導された。
CXDXXXXYXCXCのアミノ酸配列はアポトーシスを誘導する新たなアミノ酸コンセ ンサスシークエンスと考えられた
3
第一章
第一章 緒言緒言
第1節 背景 1.アポトーシス
アポトーシス(apoptosis)は花びらや枯れ葉が自然に落ちることを意味する ギリシャ語であり、プログラムされた細胞死を指す。アポトーシスを起こして いる細胞には、細胞に対する侵襲が原因となるネクローシス(necrosis)の細 胞から区別される形態上の特徴がある[1]。核の凝集や細胞の断片化が観察され、
細胞膜では細胞内側に分布するフォスファチジルセリンが外側に表出する。ア ポトーシスは多細胞生物の形態形成、組織の維持、生殖などに必須の現象であ る。オタマジャクシの尻尾の吸収や、自己抗原に反応する免疫細胞の除去など、
広範な生理的現象で観察される[2]。また臨床医学的にも重要であり、がん細胞 やウイルス感染細胞などの病的な細胞や、紫外線、放射線、熱傷、虚血などの ストレスにより障害された細胞はアポトーシスによって除去される[3]。
アポトーシスの情報伝達経路には、ミトコンドリアが関与する内因性と、Fas をはじめとする細胞死受容体を介する外因性の二種類がある[4]。内因性経路は、
紫外線などによりDNAが障害されたり、虚血などでミトコンドリアが障害された りすると活性化される。障害を受けたミトコンドリアからはSmac、AIF、HtrA2 など多くのアポトーシス誘導タンパクが放出される。中でも重要なのはチトク ロームCで、イニシエーターカスパーゼのカスパーゼ9の活性化を介し、エフェ クターカスパーゼであるカスパーゼ3を活性化する。エフェクターカスパーゼに よりアポトーシスの情報が核内に伝達される。一方、外因性経路は、FasLやTNF
(tumor necrosis factor)などの刺激を受けて活性化される。それぞれが、Fas
4
やTNF受容体に結合すると、Fas-associated death domain protein(FADD)や TNF receptor-associated death domain protein(TRADD)とカスパーゼ8や10 の複合体が形成される。活性化されたカスパーゼ8や10がカスパーゼ3を活性化 し、アポトーシスが誘導される。内因性経路でも外因性経路でも、エフェクタ ーカスパーゼであるカスパーゼ3や7の活性化がアポトーシスの誘導のための 共通経路となっている[5]。
2.EGFモチーフ
上皮成長因子(epidermal growth factor; EGF)モチーフは、約40個のアミノ 酸から構成され、その名の由来であるEGFとホモロジーをもつモチーフである[6, 7, 8]。EGFモチーフは6個のシスチン残基を含み、その配列により特徴付けられ るコンセンサスシークエンスである(図1)。二つのシスチン残基間でのジスル フィド結合により三つのループが形成される。真ん中のループBはアミノ酸配列 が最も変化に富む部位で、受容体への選択性に関わると考えられている。この モチーフは600以上の細胞外タンパクにより共有される。多くの場合、一つのタ ンパクの中に数個から数十個のEGFが連続的に存在する。EGFモチーフの約25%
がカルシウム結合性である。カルシウム結合性EGFは細胞外のカルシウムプール 中最大であり、生物学的な重要性が推察される。カルシウム結合性EGFモチーフ の多くがCX(D/N)XXXX(F/Y)XCXCのコンセンサスシークエンスを有している(Xは 任意のアミノ酸残基を表す)。D(アスパラギン酸)残基あるいはN(アスパラギ ン)残基がβ水酸化を受けるが、その機能的意義はわかっていない。
KitanoらはCX(D/N)XXXX(F/Y)XCXCのアミノ酸配列で定義されるEGFモチーフ のサブファミリーとしてCXDXXXXYXCXCを提案している[9]。彼らは、血管内皮細 胞が分泌する細胞外基質タンパクであるDel1の第三EGFドメインについて研究
5
した。Del1の第三EGFドメインのアミノ酸配列はCVDLGNSYLCRCであり、
CX(D/N)XXXX(F/Y)XCXCの条件を満たしている(図2)[10]。Del1の第三EGFドメ インは細胞のエンドサイトーシスを介する遺伝子導入の効率を改善する。この 活性は、DをNに置換したりY(チロシン)をF(フェニルアラニン)に置換した りすると消失するため、CXDXXXXYXCXCはCX(D/N)XXXX(F/Y)XCXCのなかでサブフ ァミリーを構成すると考えられた。遺伝子導入効率改善機能に加えて、Del1の 第三EGFドメインにはアポトーシス誘導作用があることが報告されている[11, 12]。CXDXXXXYXCXCのアミノ酸配列をもつタンパクには、Del1の他に、血液凝固 に関わる凝固第VII、IX、X因子や、NotchとそのリガンドであるJaggedとDLLが 挙げられる。Kitanoらは、凝固第IX因子(以下、F9と呼ぶ)の第一EGFドメイン には、遺伝子導入効率を改善する機能以外に、細胞接着を抑制する作用がある と報告した[13]。凝固第VII因子と第X因子のEGFドメインも同様な活性を示した。
これらの結果はCXDXXXXYXCXCがアミノ酸配列のみならず機能的にもファミリー を構成することを示唆している。
3.凝固第IX因子(coagulation factor IX; F9)
血液凝固は多くの物質の連鎖的反応により達成される(図3)。F9はその一つ であり、F9遺伝子の異常は血友病Bの原因となることが知られている[14, 15]。
マウスのF9は471個のアミノ酸からなる血漿タンパクであるが、翻訳後にN末端 が切断されるため、血漿中に存在するのは47番目から471番目のアミノ酸残基か らなるタンパクである。このタンパクは、N末端側の軽鎖、C末端側の重鎖と、
それらをつなぐ活性化ペプチドから構成されている。軽鎖と重鎖は、活性化ペ プチド以外にアミノ酸間のジスルフィド結合でも繋がっている(図4)。凝固反 応の過程で、F9は活性化凝固第VII因子と組織因子の複合体や活性化凝固第XI因
6
子により二箇所で限定分解される。こうして活性化ペプチドが切り離され、ジ スルフィド結合で連結された軽鎖と重鎖の二量体である活性化F9となる。
活性化F9の重鎖はタンパク分解酵素活性を持ち、凝固第X因子を限定分解によ り活性化して、凝固反応を進める。一方、F9の軽鎖は三つのドメインから構成 される(図4)。N末端側のGlaドメインは、活性化血小板膜上のフォスファチジル セリンへの結合能がある。また、IV型コラーゲンとの結合能もあり、F9の血管 内皮細胞表面への分布に寄与する。軽鎖のC末端側には二つのEGFドメインが並 ぶ。第一EGFドメインはCKDDISSYECWCのアミノ酸配列を含み、前述の
CXDXXXXYXCXC配列を持つEGFモチーフに該当する。第一EGFドメインの異常は重 鎖のタンパク分解酵素活性の低下を引き起こし、F9の凝固因子としての活性に 必須である[16]。また、Glaドメインの安定性にも寄与している[17]。第二EGF ドメインはvon Willebrand因子結合ドメインであり、止血凝固に必須の役割を 果たす。こうしたF9の構造や活性化のプロセスは、F9と同じビタミンK依存性凝 固因子である凝固第VII、X因子にもほぼ共通している(図5)。
凝固因子は、凝固止血のためのタンパクとして考えられてきた。近年、幾つ かの凝固因子には凝固活性以外に細胞への機能があることが判明し、その生理 学的・病理学的意義についての研究が増えている。例えば、トロンビンは血管 内皮細胞の接着を抑制し、血管透過性を亢進させると報告されている[18, 19]。
トロンビンのこれらの機能は敗血症や動脈硬化などの重要疾患の病態に深く関 係しており、それに関連する研究の重要度が増している。F9の機能について行 われる本研究は、F9のみならずホモロジーのあるEGFモチーフを持つ凝固因子の 機能にも関連する可能性があり、凝固系と細胞の関係についての理解を深める と期待される。
7
第2節 本研究の目的
本研究の目的は、F9の第一EGFドメイン(以下、EGF-F9と呼ぶ)の細胞への機 能を明らかにすることである。特にアポトーシス誘導能について検討を加える。
さらに、CXDXXXXYXCXCのアミノ酸配列を持つEGFドメインの医学的意味について 考察する。
8
第二章
第二章 対象と方法対象と方法
第1節 細胞と培養
実験にはヒト扁平上皮癌細胞由来のA431NS細胞(ATCC、Manassas、VA、USA)
を用いた。培養には64培養液を使用した。64培養液は、Opti-MEM I Reduced Serum 培養液(Life Technologies、New York、NY、USA)とLHC-8培養液(Life Technologies)
を6:4に混合して作製した。A431NS細胞は64培養液を用いて37℃、5%CO2気相下 で培養した。細胞が飽和状態になった時点で、細胞をTrypsin/EDTA(Lonza、
Walkersville、MD、USA)で剥がし、0.05%Trypsin Neutralizing Solution(Lonza)
でTrypsin/EDTAの活性を中和した後、1050rpmで5分間遠心した。細胞を64培養 液に再浮遊させ、継代培養を行った。
第2節 試薬
ヒト血漿由来の血液凝固第IX因子(F9)と活性化血液凝固第IX因子(以下、
活性化F9と呼ぶ)はThermo Scientific社(Waltham、MA、USA)から購入した。
マウスF9欠失変異体のcDNA(アミノ酸残基47-471、47-236、47‐191、97‐130)
を逆転写ポリメラーゼ反応(Reverse Transcription Polymerase Chain Reaction; RT−PCR)により作製し、発現ベクターであるAP-tag4 ベクター
(GenHunter、Nashville、TN、USA)に挿入した。これらのプラスミドをChinese Hamster Ovary細胞に導入し、培養することにより、組み換えタンパクを作成し た。組み換えタンパクはそれぞれ、アミノ酸残基47-471, 47-236, 47-191, 97-130
(EGF-F9)とし、コントロールタンパクをAP-tag4とした。組み換えタンパクの 濃度を測定するためにAPの発色反応を利用した。20μlの組み換えタンパクの入 った培養液に対し200μlの発色溶液(1 mg/ml p-nitrophenyl phosphate 、1 mM
9
MgCl2、1M diethanolamine、pH 9.8)を添加して、室温で30分反応させ、Smart Spec Plus分光光度計(Bio-Rad、Hercules、CA、USA)を用いて405nmでの吸光 度を測定した。Kitanoらの報告を参考とし、十分な反応をえられる濃度として 100nMの組み換えタンパクを細胞培養液に添加して実験を行った[9, 11]。
免疫染色には、ウサギ抗Paxillin抗体(Abcam、Cambridge、MA、USA)、ヤギ 抗ウサギIgG-AlexaFluor488(Life Technologies) または、ヤギ抗ウサギIgG AlexaFluor568 (Life Technologies)を使用した。対比染色に
phalloidin-AlexaFluor488(Life Technologies)または、
phalloidin-AlexaFluor568(Life Technologies)とHoechst 33342(Dojindo、
Kumamoto、Japan)を使用した。
ウェスタンブロットにはブロッディング液としてTrizma base
(Sigma-Aldrich、St.Louis、MO、USA)、メタノール(Sigma-Aldrich)、
6-Aminohexanoic Acid Cica-Reagent(KANTO CHEMICAL CO INC、Tokyo、Japan)
を使用した。ブロッキング液にはEz Blok Chemi(ATTO)を使用した。一次抗体 として、ウサギ抗Cleaved Caspase-3 (Asp175)抗体(Cell Signaling Technology、
Danvers、MA、USA)、ウサギ抗Caspase-3 抗体(Cell Signaling Technology)、
ウサギ抗Phospho-p38MAP Kinase (Thr180/Tyr182)抗体(Cell Signaling Technology)、ウサギ抗p38MAPK(D13E1)XP抗体(Cell Signaling Technology)、
ウサギ抗Phospho-p44/42MAPK(Thr202/Tyr204)(D13.14.4E)XP抗体(Cell Signaling Technology)、ウサギ抗Phospho-SAPK/JNK(Thr183/Tyr185)(81E11)抗 体(Cell Signaling Technology)、ウサギ抗β-Actin(13E5)抗体(HRP Conjugate)
(Cell Signaling Technology)、二次抗体として、ウサギ抗IgG HRP標識抗体(Cell Signaling Technology)を使用した。抗体希釈するためWestern BLoT Immuno Booster Solution1(Takara Bio、Shiga、Japan)、Western BLoT Immuno Booster
10
Solution2(Takara Bio)を使用した。発光反応のためWestern Blot Ultra Sensitive(Takara Bio)を使用した。カスパーゼ3阻害剤は、Caspase-3 InhibitorV(Merk Millipore、Darmstadt、Germany)とCaspase-3 Inhibitor VII(Merk Millipore)を使用した。
第3節 細胞形態の観察
組み替えEGF−F9タンパクがA431NS細胞の細胞外基質への接着に及ぼす影響を 検討した。直径30㎜のプラスティックディッシュ(IWAKI、Tokyo、Japan)にカ バーガラス(MATSUNAMI、Osaka、Japan)を敷き、培養皿の底面を10%ファイブ ロネクチン溶液(Sigma-Aldrich)でコートし、A431NS細胞を撒いて60分間培養 した。播種細胞数は10×10⁴cell/mlとした。その後タイムラプス画像撮影装置 CellWatcher(Corefront Corporation、Tokyo、Japan)を用いて連続観察した。
サンプル数はn=15とした。AP-tag4で処理したものをコントロールとした。
細胞の接着面積の測定を行った。直径30㎜のプラスティックディッシュにカ バーガラスを敷き、培養皿の底面を10%ファイブロネクチン溶液でコートし、
A431NS細胞を撒いて60分間培養した。播種細胞数は10×10⁴cell/mlとした。100 nMのEGF−F9を加えて10分間培養後に4%パラフォルムアルデヒド(Wako)溶液で 細胞を固定した。細胞膜をDi-I(Life Technologies)で染色後、共焦点顕微鏡
(Leica、Wetzlar、Germany)を使用してガラス面の細胞膜を撮影した。ガラス 面の細胞膜面積を画像解析ソフトウェアPopImaging(Digital Being Kids、
Kanagawa、Japan)を用いて計測した。サンプル数はn=20とした。AP-tag4で処 理したものをコントロールとした。結果は平均±標準偏差で表した。
第4節 免疫細胞化学染色
11
細胞内の接着分子の局在を検討するために免疫細胞化学染色を行った。
Microplate-24 well(IWAKI)にカバーガラスを敷きファイブロネクチンでコー トしてA431NS細胞を捲き、60分間培養した。播種細胞数は0.5×10⁵cell/mlとし た。EGF-F9を100nM加えたものをEGF-F9群とした。EGF-F9存在下で10分間培養 し、培養液を除去した。その後、4%パラフォルムアルデヒド溶液で細胞を固定 した。次に、0.1%TritonX-100 Solution(Sigma-Aldrich)/リン酸緩衝生理食 塩(Phosphate buffered saline; 以下PBSと呼ぶ)にて室温で5分間処理し、3%
牛血清アルブミン(Bovine serum albumin; BSA)/PBSで1時間ブロッキングし た。PBSで1:1000に希釈したウサギ抗Paxillin抗体を4℃で16時間反応させ、洗 浄後に1:1000に希釈したヤギ抗ウザギIgG-Alexa Fluor488を室温で60分反応さ せた。対比染色は、アクチンの染色にphalloidin-AlexaFluor568 、核の染色に Hoechst 33342を用いて行った。洗浄・封入後、蛍光顕微鏡(Carl Zeiss、Welwyn Garden City、UK)で観察した。
第5節 細胞接着の評価
細胞基質間接着の強度を評価するために以下の実験を行った。Microplate-96 well(IWAKI)を10%ファイブロネクチン溶液でコートした。A431NS細胞を64培 養液中で60分間培養した。播種細胞数は0.5×10⁴cell/mlとした。その後、F9、
活性化F9、47-471、47-236、47-191およびEGF-F9を100nM培養液中に添加した。
タンパク添加後に、培養皿を室温で30分間、Plate Mixer(Asone、Osaka、Japan)
上で(1050rpm)振盪した。培養液を回収し、底面より剥がれた細胞数を血球計算 盤(ERMA、Tokyo、Japan)を用いて顕微鏡下で計測した。サンプル数はn=5とし た。結果は、コントロールで剥がれた細胞数の平均を1として、平均±標準偏差 で表した。
12
第6節 Lactate dehydrogenase(LD)活性測定
細胞死の量を評価するために培養液中のLD活性を測定した。LD測定には、細胞傷 害性検出キットplus LDH Version06(Roche Diagnostics、Tokyo、Japan)を使 用した。細胞傷害性の割合を計算するために3種類のコントロールを用意した。
培養液中に含まれるLD活性を測定しバックグラウンドコントロールとした。ま た、未処理の正常細胞から放出されるLD活性を測定し低コントロール、付属の 細胞溶解バッファーを使用し細胞中の最大LD活性を測定し高コントロールとし た。本研究では、64培養液のみをバックグラウンドコントロール、64培養液に A431NS細胞を加えたものを低コントロール、64培養液にA431NS細胞を加えさら に細胞溶解バッファーを加えたものを高コントロールとした。
Microplate-96 well にA431NS細胞を蒔き60分間培養した。播種細胞数は 0.5×10⁴cell/mlとした。その後100nMのEGF−F9を添加し24時間後に培養液を回 収して、ELISAリーダー( TECAN、Kanagawa、Japan)を使用し室温下で各々の LD活性を測定した。サンプル数はn=5とし、実験は3回行った。AP-tag4で処理し たLD活性の平均を1として、平均±標準偏差で表した。
第7節 アネキシンV染色
アポトーシスをおこした細胞数を知るため、アネキシンV染色を行った。直径 35㎜のGlass Bottom Dish(MATSUNAMI)をファイブロネクチンでコートし、A431NS 細胞を撒いて60分間培養した。播種細胞数は0.5×10⁵cell/mlとした。EGF-F9を 100nM加えたものをEGF-F9群とし、AP-tag4を加えたものをコントロールとした。
EGF-F9存在下で8時間培養した細胞に、培養液量の5%の量のアネキシンV
13
AlexaFluor 568(Invitrogen、Carlsbad、CA、USA)を加え、遮光下に室温で15 分間反応させた。4%パラフォルムアルデヒドで固定後、0.1% Triton X-100/PBS で室温にて5分間処理した。phalloidin-AlexaFluor488を用いてアクチンの染色 を行った。その後、Leica社の共焦点顕微鏡で観察し写真を撮影した。撮影した 画像の全細胞数とアネキシンV染色陽性細胞数を数え、アネキシンV染色陽性細 胞数の割合を計算した。サンプル数はn=5とし実験は3回行った。結果は平均±
標準偏差で表した。
第8節 ウェスタンブロット法によるEGF-F9の影響の評価
細胞が産生するタンパク量を評価するためウェスタンブロットを行った。A431NS細胞 をファイブロネクチンでコートしたMicroplate 24wellに撒き、1時間培養後に EGF−F9を100nmol加えたものをEGF-F9群とし、AP-tag4を加えたものをコントロ ールとした。播種細胞数は、0.5×10⁵cell/mlとした。EGF-F9で処理し、時間を 追って100μlのサンプルバッファー[1% sodium dodecyl sulfate(Wako、Osaka、
Japan)、1% 2-mercaptoethanol(Wako)、50mM Tris-HCl(Sigma-Aidrich)pH6.8、
20% glycerol(Wako)、bromophenol blue(Wako)]を加えてサンプルを作製し た。超音波処理後、100℃で5分間処理し、-20℃で保存した。
泳動槽(ATTO、Tokyo、Japan)に泳動槽用緩衝液[25mM Tris-base(Wako)、
192mM Glycine(Wako)、0.1% sodium dodecyl sulfate)]を満たし、E・パジェ ル AE-6000シリーズ電気泳動用既製ゲル5-20%(ATTO)に15μlのサンプルを アプライし、20mAで電気泳動した。泳動終了後、セミドライ式ブロッティング 装置(ATTO)を用いて、Clear Blot Membrane膜(ATTO)に転写した。ブロッテ ィング液は、A溶液[Trizma base1M、6.5ml、メタノール0.5ml、以上に純水を 加え20mlとした]とB溶液[Trizma base1M、0.5ml、メタノール0.5ml、以上に純
14
水を加え、(HClでpH8.8に調整 )20mlとした] とC溶液[Trizma base1M、0.5ml、
メタノール0.5ml、6-Aminohexanoic Acid Cica-Reagent 0.1g、以上に純水を加 え、(HClでpH6.8に調整)20mlとした]を使用した。
蒸留水で5倍に希釈したEz Blok Chemiを用い、転写した膜を室温で30分間ブ ロッキングした。
一次抗体をWestern BLoT Immuno Booster Solution1で1000倍に希釈し、転写し た膜を浸して室温で60分、または4℃で一晩振盪した。転写した膜をPBSで3回洗 浄した後、5mlのWestern BLoT Immuno Booster Solution2で抗ウサギIgG HRP付 属抗体を1:5000に希釈した液に浸して、室温で60分振盪した。PBSで3回洗浄し た後、発光反応のためにWestern Blot Ultra Sensitiveを転写した膜に滴下し、
暗室内でHyperfilm ECL(GE Healthcare、Tokyo、Japan)に露光した。または Lumi Cube(Liponics、Tokyo、Japan)を使用して撮影した。得られたバンドの 定量化はJust TLC software(Sweday、Lund、Sweden)で行った。実験は3回行 い、結果はコントロールの平均結果を1として、平均±標準偏差で表した。
第9節 カスパーゼ3阻害剤存在下でのLD活性測定
EGF-F9によるアポトーシスのカスパーゼ 3依存性について検討するために、
培養液中の LD活性を測定した。測定には、細胞傷害性検出キット plus LDH Version06を使用した。第 6節と同様に 3種類のコントロールを用意した。
Caspase-3 InhibitorVと Caspase-3 Inhibitor VIIを DMSOで希釈し 100nMとし た。Microplate-96 well に A431NS細胞を蒔き 60分間培養した。播種細胞数は 0.5×10⁴cell/mlとした。各々のカスパーゼ 3阻害剤で処理し 30分後 100nMの EGF−F9を添加し 24時間後に培養液を回収して、TECAN社の ELISAリーダーを使 用し室温下で各々の LD活性を測定した。サンプル数は n=5とし実験は 3回行っ
15
た。コントロールの LD活性の平均を 1として、平均±標準偏差で表した。
第10節 カスパーゼ3阻害剤存在下でのアネキシンV染色
EGF-F9によるアポトーシスのカスパーゼ3依存性について検討するために、ア ネキシンVで染まる細胞数を測定した。直径35㎜のGlass Bottom Dish(MATSUNAMI)
をファイブロネクチンでコートし、A431NS細胞を撒いて60分間培養した。播種 細胞数は0.5×10⁵cell/mlとした。Caspase-3 InhibitorVをDMSOで希釈し100nM とした。カスパーゼ3阻害剤で処理し30分後EGF-F9を100nM加えたものをEGF-F9 群とし、AP-tag4を加えたものをコントロールとした。EGF-F9で処理し、8時間 培養した細胞に、培養液量の5%の量のアネキシンVを加え、遮光下に室温で15 分間反応させた。4%パラフォルムアルデヒドで固定後、0.1% Triton X-100/PBS で室温にて5分間処理した。phalloidin-AlexaFluor488を用いてアクチン染色を 施した。Leica社の共焦点顕微鏡で観察し写真を撮影した。撮影した画像上の全 細胞数とアネキシンV染色陽性細胞数を数え、アネキシンV染色陽性細胞数の割 合を計算した。サンプル数はn=5とし実験は3回行った。結果は平均±標準偏差 で表した。
第11節 活性化カスパーゼ3陽性細胞の評価
カスパーゼ3活性化の時間経過を評価するために、細胞の活性化カスパーゼ3を染 色した。Microplate-24 wellにカバーガラスを敷きファイブロネクチンでコート してA431NS細胞を捲き、60分間培養した。播種細胞数は0.5×10⁵cell/mlとした。
EGF-F9を100nM加え5、10、30分間培養した。コントロールにはAP-tag4を使用 した。培養液を除去後、4%パラフォルムアルデヒド溶液で細胞を固定した。次 に、0.1%TritonX-100 Solution/PBSにて室温で5分間処理し、3%BSA /PBS で1
16
時間ブロッキングした。PBSで1:1000に希釈したウサギ抗Cleaved
Caspase-3(Asp175)抗体を4℃で16時間反応させ、洗浄後に1:1000に希釈したヤ ギ抗ウザギIgG-Alexa Fluor488を室温で60分反応させた。対比染色は、核の染 色にHoechst 33342を用いて行った。洗浄・封入後、観察はCarl Zeissの蛍光顕 微鏡で行った。
第12節 接着阻害培養
接着の阻害によりカスパーゼ3の活性化がおこるか検討するために、ウェスタンブロッ トを行った。Trypsin/EDTAを用いてA431NS細胞を剥がし、0.05%Trypsin
Neutralizing Solutionで中和した後に遠心し、64培養液で細胞浮遊液を作製し た。細胞が底面に接着しないようにMicroplate 24wellのウェルの底にカバーガ ラスを敷いて、細胞浮遊液を入れた。播種細胞数は、0.5×10⁵cell/mlとした。
培養器中で培養しながら時間を追って細胞を回収し、遠心して得た細胞塊にサ ンプルバッファーを加えてサンプルを作製した。超音波処理後、100℃で5分間 処理し、-20℃で保存した。作成したサンプルはウエスタンブロット法にて評価 した。実験は3回行った。
第13節 カスパーゼ3阻害剤存在下での細胞接着の評価
カスパーゼ3阻害剤存在下での細胞基質間接着の強度を評価するために以下の 実験を行った。Microplate-96 wellを10%ファイブロネクチン溶液でコートした。
A431NS細胞を64培養液中で60分間培養した。播種細胞数は0.5×10⁴cell/mlとし た。Caspase-3 InhibitorVとCaspase-3 Inhibitor VIIをDMSOで希釈し100nMと した。その後、EGF-F9を100nM培養液中に添加した。タンパク添加後に、培養 皿を室温で30分間、Asone社のPlate Mixer上で(1050rpm)振盪した。培養液を回
17
収し、底面より剥がれた細胞数を血球計算盤を用いて顕微鏡下で計測した。サ ンプル数はn=5とした。結果は、コントロールで剥がれた細胞数の平均を1とし て、平均±標準偏差で表した。
第14節 カスパーゼ3阻害剤存在下でのMitogen-activated protein kinase (MAPK)のタンパク産生の検出
EGF-F9によるMAPKリン酸化のカスパーゼ3依存性を検討するために、ウェスタンブ ロットを行った。Caspase-3 InhibitorVをDMSOで希釈し100nMとした。A431NS細胞を ファイブロネクチンでコートしたMicroplate 24wellに撒き、60分間培養後カス パーゼ3阻害剤で処理し30分後、EGF-F9を100nM加えたものをカスパーゼ3阻害 剤存在下EGF-F9群とし、カスパーゼ3阻害剤を使用せずEGF−F9を100nMを加えた ものをEGF-F9群とした。AP-tag4を加えたものをコントロールとした。処理後30 分でサンプルバッファーを加えてサンプルを作製した。超音波処理後、100℃で 5分間処理し、-20℃で保存した。実験は3回行った。
作成したサンプルはウエスタンブロット法にて評価した。得られたバンドの 定量化はJust TLC softwareで行った。
第15節 統計分析
Mann-WhitneyのU検定を行い、検定結果の指標はp値を用いた。95%信頼区間の 場合は p < 0.05 、99%信頼区間の場合は p < 0.01 をもって有意差があると判 断し、図中ではそれぞれ*、**で表した。結果は平均±標準偏差とした。
18
第三章
第三章 結果結果
第 1節 細胞接着への影響
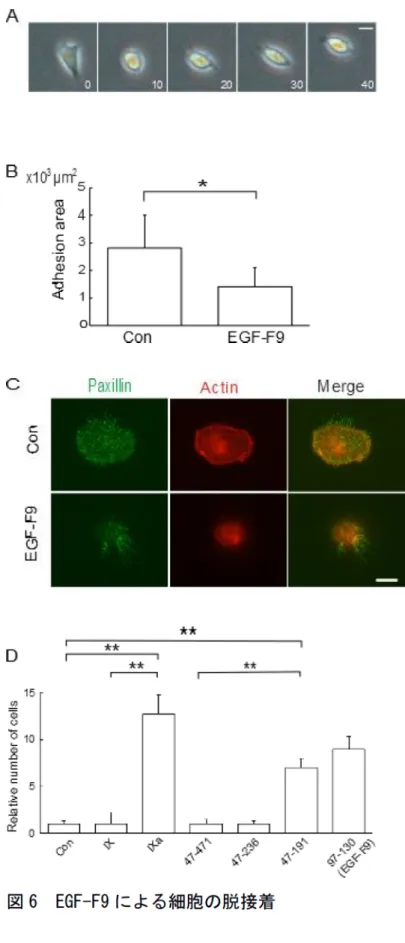
ファイブロネクチンでコートした培養皿に接着した細胞に100 nMのEGF−F9を 添加し、顕微鏡下で観察した(図6A)。細胞は刺激から10分以内に星型から球形 になった。この状態で培養皿を軽くゆすっても細胞は動かないため、完全な細 胞脱接着は生じていなかった。しかし、細胞の形態から細胞と基質との接着面 積の低下が推察された。そこで、細胞膜を蛍光色素Di-Iで染色し、共焦点顕微 鏡で細胞基質接着部の面積を測定したところ、EGF−F9で処理した細胞の接着部 面積は、刺激から10分後に50%減少していることが確認された(図6B)。次に、
細胞基質間接着部での接着分子の状態を調べるため、パキシリンに対する抗体 とアクチン結合タンパクであるファロイジンを利用して免疫染色をおこなった
(図6C)。コントロールの細胞では重合したアクチン繊維が細胞辺縁に沿って円 形に染色され、パキシリンがそれと共存し、接着装置が形成されていた。EGF−F9 を培養液に添加すると、アクチンもパキシリンも減少して円形の分布は消失し、
フィロポディアへの分布を残すだけになった。これらの結果より、EGF−F9は極 めて短時間に接着斑の形成を阻害し、細胞基質間接着を抑制することが分かっ た。
EGF−F9の細胞接着に関する機能が凝固反応とどのように関係しているか検討 するため、F9タンパク、活性型F9タンパクおよびF9の欠失変異体を用いて実験 を行った。培養皿に蒔いたA431NS細胞にタンパクを添加後、培養皿をシェイカ ー上で30分振とうし、剥がれてきた細胞数を血球計算盤で計測した(図6D)。コ ントロールでは細胞はほとんど剥がれなかった。ヒト血漿由来F9タンパクおよ びマウスF9全長組み換えタンパクによる処理では細胞の剥離は誘発されなかっ
19
たが、ヒト血漿由来活性型F9タンパクによる細胞剥離が観察された。F9の欠失 変異体を用いた実験では、活性化ペプチドを含むタンパクには活性がなかった が、軽鎖あるいはEGF−F9には活性があった。
第2節 アポトーシスの誘導
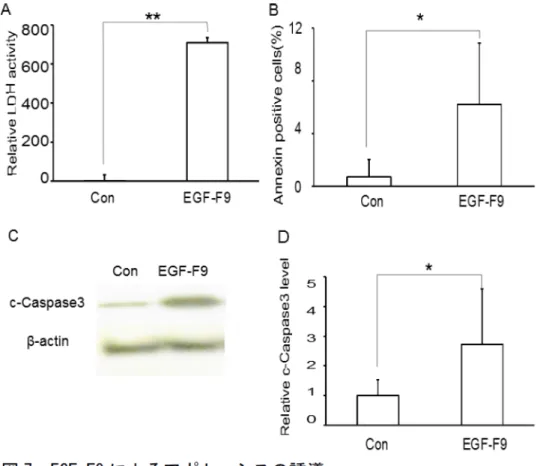
次にEGF−F9が、ホモロジーのあるDel1の第三EGFドメイン同様にアポトーシス を誘導するか検討した。細胞死の指標として培養液中のLD活性を調べた。また アポトーシスのマーカーとして、細胞表面のフォスファチジルセリンをアネキ シンVで染色し、活性型カスパーゼ3タンパク量をウェスタンブロットで検出し た。培養液中のLD活性は100 nMのEGF−F9を添加してから24時間後にコントロー ルの平均の約755倍と有意に増加した(図7A)。アネキシンVで染色される細胞は、
EGF−F9添加後8時間で評価し、EGF−F9処理細胞ではコントロールの0.7±1.3%に 対して6.2±4.6%に増加した(図7B)。活性型カスパーゼ3はEGF−F9投与後30分 で増加し、コントロールの平均の約2.7倍になった(図7C、D)。
第3節 情報伝達のカスパーゼ3依存性
活性型カスパーゼ3の増加(図7C、D)は、EGF−F9によるアポトーシスがカス パーゼ3依存性であることを示唆していた。それを確認するために、カスパーゼ 3阻害剤を用いた実験を行った。細胞死の指標であるLDの放出はインヒビターV によりコントロールの約525倍となり、EGF-F9に比べ約31±4.6%抑制された。
また、インヒビターVIIによりコントロールの約445倍となり、EGF-F9に比べ約 42±17.1%抑制された。(図8A)。次に、アネキシンVによる細胞表面のフォスフ ァチジルセリンについて検討したところ、インヒビターVによりアネキシンVで 染まる細胞はコントロールの約2.3倍となり、EGF-F9に比べ約73±36.3%抑制さ
20
れた(図8B)。しかしインヒビターVIIは、それ自体に細胞表面へのアネキシンV の結合を増やす作用があるため評価できなかった。
図6Aに示したように、EGF−F9の投与は10分以内に細胞接着を減弱する。この 結果は、EGF−F9によるアポトーシスが接着阻害によって誘発されるアノイキス である可能性を示唆した。そこで、細胞接着の減弱とカスパーゼ3活性化の関係 について検討するため、カスパーゼ3活性化の時間経過について調べた。A431NS 細胞にEGF−F9を添加し、時間を追ってサンプルを採取した。ウェスタンブロッ トを行ったところ、活性化カスパーゼ3とカスパーゼ3は刺激後5から10分で増加 が認められた(図8C)。通常、アポトーシス誘導時のカスパーゼ3の活性化は30 分から数時間かけて起こる[20, 21]。早期のカスパーゼ3活性化を確認するため に、細胞免疫染色を行った。EGF-F9の投与後5分から活性化カスパーゼ3が染色 され、陽性細胞は徐々に増加した(図8D)。
次に、このカスパーゼ3の活性亢進の原因がEGF−F9による脱接着であるのか確 認するために、細胞をガラス面に撒くことにより接着を阻害してサンプルを作 成した。その結果、強制的に細胞接着を阻害しても30分ではカスパーゼ3の活性 化はおこらないことが判明した(図8E)。EGF−F9によるカスパーゼ3の活性化に は脱接着以外のシグナルが必要と考えられた。この早期のカスパーゼ3活性化が 細胞脱接着の上流で働いている可能性を考え、カスパーゼ3阻害剤を用いた実験 を行った(図8F)。EGF−F9により誘発される細胞接着の減弱化はインヒビターV とインヒビターVIIによりそれぞれ約72±22%と約92±10%抑制された。
第4節 MAPKのリン酸化
カスパーゼ3はアポトーシスの情報伝達路の下流で働くエフェクタータンパ クと考えられてきた。しかし本研究により、EGF−F9によるアポトーシス誘導系
21
においては上流で働く可能性が示された。そこで、アポトーシスの情報伝達路 ではたらく別の因子とカスパーゼの関係についても検討を加えた。ウェスタン ブロットによりリン酸化p38MAPK量を観察したところ、EGF−F9の投与後10分より リン酸化p38MAPKが増加していた(図9A)。MAPK44/42やSAPK/JNKのリン酸化タン パクは増えておらず、p38MAPKの反応は特異的であると思われた(図9B)。リン 酸化p38MAPK増加のカスパーゼ3依存性を調べるためにインヒビターVを使用し たところリン酸化p38MAPK増加は抑制されたので、カスパーゼ3はp38MAPKリン酸 化の上流で働いていると推察された(図9C)。
22
第四章
第四章 考察考察
本研究において、活性化F9は凝固因子として働くのみならず、ヒト扁平上皮 癌細胞株に対して脱接着作用を示すことが確認された。活性化F9は細胞基質間 接着を減弱化し、その活性は第一EGFドメイン(EGF-F9)に局在した。EGF−F9の 活性は、Kitanoらが報告しているように、凝固反応に伴う活性化ペプチドの除 去により生じると推察された(図6D)[13]。またEGF-F9には、CXDXXXXYXCXC配 列を持つDel1の第三EGFドメイン同様に、アポトーシスの誘導活性があると判明 した。
第1節 EGF-F9の情報伝達路
EGF-F9により誘導されたアポトーシスに先行してカスパーゼ3の活性化が観 察された。またアポトーシスは2種類のカスパーゼ3阻害剤により部分的に抑制 された。同様に、EGF-F9によって誘発された脱接着やp38MAPKのリン酸化もカス パーゼ3阻害剤により特異的に阻害された。これらの結果は、EGF-F9によるこれ らの反応がカスパーゼ3依存性である可能性を示唆しているが、その解明には siRNA等による詳細な実験が必要と思われる。もしカスパーゼ3活性化の下流に 脱接着が位置するなら、通常のアノイキスとは逆のシグナル伝導路の存在が考 えられる[22, 23]。
EGF-F9によるカスパーゼ3の活性化は刺激後30分以内に観察された。このよう な早期のカスパーゼ3活性化についての報告はあまり多くない。Borutaiteらは ランゲンドルフ法を用いて心筋虚血モデルを作成し、カスパーゼ3の活性を検討 した[24]。その結果、心筋の還流を止めてから30分後にカスパーゼ3の活性が増
23
加した。さらに、この現象はミトコンドリアのmitochondrial permeability transition阻害剤により抑制されたことから、ミトコンドリアからのチトクロ ームcの放出が原因と考えられた。また、ショウジョウバエを用いた実験では、
ハエの幼生に針で外傷を与えると30分以内に腸管上皮でカスパーゼ3が活性化 された[25]。この実験系でカスパーゼ3の活性を抑制するとハエ幼生は死亡した。
この結果よりカスパーゼ3の活性化は外傷ストレスからの回復に必須と思われ た。In vitroの実験系を用いた研究では、Rosadoらの報告がある[26]。血小板 ではトロンビンの刺激から1分でカスパーゼ3活性が上昇した。血小板はやや特 殊な細胞と言えるが、彼らは膵臓の細胞でもCCK-8の刺激から1分でカスパーゼ 3活性の上昇を認め、この反応がアミラーゼの分泌に必須であると報告している。
外傷に対する凝固反応や、食事に対する消化酵素の分泌は分単位の反応であり、
カスパーゼ3の迅速な活性化は生体の素早い反応に共通して起こる可能性が考 えられる。本研究ではカスパーゼ3の総タンパク量も早期から増加していた。刺 激から短時間で起こるカスパーゼ3活性の亢進は、生理的な反応において重要な 役割を果たす可能性がある。
第2節 EGF-F9の医学的意味
次に、凝固因子の一つであるF9が細胞接着を減弱化したりアポトーシスを誘 発したりする医学的意味について考察する。
第一に、凝固反応が外傷部で生じることから考えると、障害を受けた組織に アポトーシスを誘導し、エンドサイトーシスによるデブリードメントを容易に する可能性がある[27]。EGF-F9はアポトーシスを誘導するだけでなく、エンド サイトーシスを亢進させることから、EGF-F9が一連の反応をサポートするのか もしれない。
24
第二に、細胞の脱接着は創の周辺組織からの創内への細胞の遊走に必要であ る。通常、創傷の治癒には創の周囲組織から、上皮細胞、線維芽細胞、内皮細 胞、マクロファージなど多種の細胞が動員される。これらの細胞が元の位置か ら離れて創内に遊走するのにEGF-F9が役立つ可能性がある。実際、F9のノック アウトマウスでは、マクロファージの遊走が抑制され、創傷の治癒が遅れると 報告されている[28, 29]。
第三に、F9は創傷治癒に必要な細胞分化や増殖に関わっている可能性がある。
ヒドラ、カエル、ハエなどの下等動物では、組織再生の過程でアポトーシスが 観察されている[30, 31, 32]。この場合のアポトーシスは、受傷から1時間以内 にアポトーシスの過程で生じる断片化DNAを検証するTUNEL
(TdT-mediateddUTPnickendlabeling)法で検出されており、極めて素早く誘導 される。このアポトーシスは創傷治癒のために周囲の細胞が代償性増殖 (compensatory proliferation)する場合に必要であり、MAPKの活性化を伴うと 報告されている[33]。哺乳類の場合、いくつかの系列でカスパーゼ3の活性化が 細胞分化に必須である。OkuyamaらはNotchからカスパーゼ3に至る情報伝達路が 皮膚角化細胞の分化を制御していると報告した[34]。また、カスパーゼ3から p38MAPKに至る情報伝達は、血液細胞の分化に必須である[35]。さらに、カスパ ーゼ3のノックアウトマウスでは皮膚や肝臓での組織の再生が抑制された[36]。
第3節 アミノ酸配列CXDXXXXYCXC
本研究においてEGF-F9のアポトーシス誘導作用が確認されたため、アミノ酸 配列CXDXXXXYCXCを持つDel1とF9のEGFドメインは、アポトーシスの誘導という 共通の機能を持つことが明らかになった。CXDXXXXYCXCが特定の機能を持つ新規 のファミリーを構成する可能性が高まったと言える。Kitanoらは、Del1の第三
25
EGFドメインのcDNAと非ウイルスベクターを用い、ヒト扁平上皮がんのマウス移 植腫瘍に遺伝子治療を施した[12]。その結果、治療群において有意な腫瘍縮小 効果と延命効果を認めた。治療群の腫瘍組織ではアポトーシスが起きているの が観察され、治療効果は腫瘍組織のアポトーシスが原因の一つと考えられた。
Del1の第三EGFドメインの細胞に対するエンドサイトーシスの亢進、アポトーシ スの誘導作用は、浮遊細胞を除く多くの細胞に対して有効と報告されている。
アポトーシスの誘導は腫瘍治療の有効な方法の一つであり、CXDXXXXYCXC配列が 固形がんの新たな治療の開発に役立つ可能性がある。
また、CXDXXXXYCXC配列による細胞脱接着やアポトーシスは動脈硬化等の血管 病変に関わっている可能性がある。この配列は、F9だけでなく凝固第VII因子や 第X因子にも存在し、細胞脱接着を起こす。動脈壁の血管内皮細胞の脱落やアポ トーシスが動脈硬化や動脈血栓の原因になることが知られており、これらの凝 固因子の関与が推察される[37]。これまで、血栓症の治療薬は凝固能を指標に 用いられてきた。凝固因子の細胞に対する機能の発見は、動脈硬化症やそれに 伴う血栓症の予防や治療における新たなターゲットの発見と考えられる。
26
第五章
第五章 まとめまとめ
本研究ではEGF-F9がアポトーシスを起こすことを示した。EGF-F9の活性化は 凝固反応と同様のプロセスによると思われた。EGF-F9のアポトーシス誘導作用 の一部は、カスパーゼ3活性化を介するシグナル経路の関与が示唆された。
CXDXXXXYCXCのアミノ酸配列は凝固第VII因子と第X因子にも共通するため、凝固 因子としての系以外に、細胞に対する機能に関わる新たな生理学的な系が存在 する可能性がある。細胞接着やアポトーシスは創傷の治癒、組織の再生、血管 病の発症、がん治療などに関与しており、CXDXXXXYCXCのアミノ酸配列は様々な 分野で重要な研究対象になると考えられる。
27
謝辞 謝辞
本研究を遂行し学位論文をまとめるに当たり、御指導を賜りました、指導教官 である日本大学医学部生体機能医学系生理学分野 國分眞一朗 教授に深く感 謝しております。
また、研究全般にわたる多大なる御支援、御指導を賜りました 日臺智明 准 教授に御礼申し上げます。
さらに、本研究を遂行するに当たり、御協力と御理解を頂きました日本大学 医学部生体機能医学系生理学分野諸先生、スタッフの方々、さらに、御理解お よび御配慮を頂きました日本大学医学部耳鼻咽頭・頭頸部外科系歯科口腔外科 学分野 吉田美昭 講師、御指導および御助言を頂いた 北野尚孝 助教、真 宮淳 助手を始め、歯科口腔外科医局の諸先生方に深く感謝致します。
本論文作成に当たり、審査委員として多くの御助言を頂きました 中山智祥 教授、松本太郎 教授、浅井聰 教授、仲沢弘明 教授には深く感謝いたしま す。
最後に、支えとなった家族に感謝致します。
28
図 図
図1 EGFモチーフの構造
図2 アミノ酸配列の比較
29
図3 血液凝固カスケード
30
図4 マウスF9タンパクの構造
図5 ビタミンK依存性凝固因子のタンパク構造
31
図 6 EGF-F9による細胞の脱接着
32
図 7 EGF-F9によるアポトーシスの誘導
33
34
図8 EGF−F9によるアポトーシスのカスパーゼ3依存性
35
図9 EGF−F9によるリン酸化MAPKの変化
36
図10 創傷治癒の過程
37
図説 図説
図1 EGFモチーフの構造
EGFモチーフの構造模式図。丸はアミノ酸残基を、黒丸はシスチン残基を表す。
シスチン同士をつなぐ線はジスルフィド結合を表す。
図2 アミノ酸配列の比較
マウスのビタミンK依存性凝固因子の第一EGFドメインとDe11第三EGFドメイ ンのアミノ酸配列を比較した。共通するアミノ酸を枠で囲った。数字はマウス 凝固第IX因子のアミノ酸番号。C;シスチン、K;リジン、D;アスパラギン酸、
I;イソロイシン、S;セリン、Y;チロシン、E;グルタミン酸、W;トリプトフ ァン、Q;グルタミン、H;ヒスチジン、L;ロイシン、V;バリン、F;フェニル アラニン、G;グリシン、T;スレオニン、N;アスパラギン、R;アルギニン。
図3 血液凝固カスケード
凝固反応の概要を図に表した。青は内因系、赤は内因系、緑は共通系の反応 を示す。
38
図4 マウスF9タンパクの構造
マウスF9タンパクは471個のアミノ酸からなるタンパクとして翻訳され、血液 中に分泌される前にN末端のリーダー配列(Pre、Pro)が切除される。軽鎖と重 鎖は活性化ペプチドで繋がる以外に、アミノ酸間のジスルフィド結合(S−S)に よって二量体になっている。軽鎖はGla、第一EGFドメイン(EGF−1)、第二EGFド メイン(EGF−2)の三つのドメインから構成される。凝固因子としてのタンパク 分解酵素活性は重鎖に存在する。
図5 ビタミンK依存性凝固因子のタンパク構造
ビタミンK依存性凝固因子の構造を図に示した。いずれもN末端にフォスファ チジルセリンに結合するGlaドメインを持ち、C末端にタンパク分解酵素活性が ある。A; A chain、CR; connecting region、AP; activating peptide。
図6 EGF-F9によるA431NS細胞の脱接着
A)細胞形態の変化。数字は時間(分)を示す。スケールバーは20μmを示す。
B)100 pmol EGF-F9投与10分後の細胞接着面積の測定。コントロール(Con)
にはAP-tagタンパクを使用した。
n=15。
C)パキシリン(Paxillin)とアクチン(Actin)の免疫染色。コントロール
(Con)にはAP-tagタンパクを使用した。スケールバーは10μmを示す。
D)脱接着細胞の測定。横軸の数値はF9変異体のアミノ酸残基数を示す。47-471 がF9全長、97−130がEGF−F9に相当する。n=6。
図 7 EGF-F9によるアポトーシスの誘導
39
A)24時間後培養液中に放出されたLD活性。コントロール(Con)にはAP-tag タンパクを使用した。n=5。
B)8時間後のアネキシンV染色陽性細胞の割合。n=6。
C)30分後の活性化カスパーゼ3(c-Caspase3)のウェスタンブロット。
D)C)のウェスタンブロットの発光強度を測定した。n=3。
図8 EGF−F9によるアポトーシスのカスパーゼ3依存性
A)EGF-F9により培養液中に放出される24時間後のLD活性に対するカスパーゼ 3阻害剤[DMSO(Vehicle)、インヒビターV(InhV)、インヒビターVII(InhVII)]
の効果。コントロール(Con)にはAP-tagタンパクを使用した。n=3。
B)8時間後のアネキシンV染色陽性細胞の割合に対するインヒビターV(InhV)
の効果。コントロール(Con)にはAP-tagタンパクを使用した。
n=3。
C)EGF-F9によるカスパーゼ3(Caspase3)と活性型カスパーゼ3(c-Caspase3)
の発現をウェスタンブロットで評価した。コントロール(Con)にはAP-tagタン パクを使用した。βアクチンはサンプル量のコントロール。
D)EGF-F9による活性型カスパーゼ3(c-Caspase3)の陽性細胞を免疫染色し検 証した。コントロール(Con)にはAP-tagタンパクを使用した。Hoechst33342に よる核染色結果は画像撮影後、疑似カラーで表現した。スケールバーは50μmを 示す。
E)強制的な脱接着による活性型カスパーゼ3(c-Caspase3)の発現。細胞が 接着できないガラス底の培養皿に蒔いたもので強制的な脱接着サンプルを作製 した。同時に、接着細胞(Con)と接着細胞にEGF−F9を作用させたサンプル(EGF-F9)
を用意した。βアクチンはサンプル量のコントロール。
40
F)EGF−F9により誘導された脱接着に及ぼすインヒビターV(InhV)とインヒ ビターVII(InhVII)の影響。VehicleとしてDMSOを使用。図5Dと同様の実験を 行い、EGF−F9存在下で振盪を加えた時に生じる細胞剥離へのカスパーゼ3阻害剤 の影響を調べた。n=5。
図9 EGF−F9によるリン酸化MAPKの変化
A)EGF-F9によるリン酸化p38MAPK(p-p38MAPK)の発現。A431NS細胞に 100pmol/mlのAP-tag付きEGF-F9(EGF−F9)を投与した。経時的にサンプルを採 取し、ウエスタンブロッティングを行った。βアクチンはサンプル量のコント ロール。
B)EGF-F9によるリン酸化MAPK44/42(p-MAPK44/42)とリン酸化SAPK/JNK
(p-SAPK/JNK)の発現。
C)EGF-F9によるリン酸化p38MAPK(p-p38MAPK)の発現に対するDMSO(Vehicle)
とインヒビターV(InhV)の影響。
図10 創傷治癒の過程
障害を受けた組織には血液や周辺組織からの創内への上皮細胞、線維芽細胞、
内皮細胞、マクロファージなどの細胞が動員される。それらの細胞が分化、増 殖して創傷治癒を進める。
41
引用文献 引用文献
[1] ELMORE S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495-516.
[2] DEBNATH J. Detachment-induced autophagy during anoikis and lumen formationin epithelial acini. Autophagy 2008; 4: 351-353.
[3] GUADAMILLAS M C, CEREZO A and DEL POZO M A. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J Cell Sci 2011; 124: 3189-3197.
[4] PORTT L, NORMAN G, CLAPP C, GREENWOOD M and GREENWOOD M T. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta 2011; 1813: 238-259.
[5] SHALINI S, DORSTYN L, DAWAR S and KUMAR S. Old, new and emerging functions of caspases. Cell Death Differ 2015; 22: 526-539.
[6] WINSHIP P R and DRAGON A C. Identification of haemophilia B patients with mutations in the two calcium binding domains of factor IX: importance of a beta-OH Asp 64----Asn change. Br J Haematol 1991; 77: 102-109.
[7] WOUTERS M A, RIGOUTSOS I, CHU C K, FENG L L, SPARROW D B, et al. Evolution of distinct EGF domains with specific functions. Protein Sci 2005; 14: 1091-1103.
[8] STENFLO J, STENBERG Y and MURANYI A. Calcium-binding EGF-like modules in coagulation proteinases: function of the calcium ion in module interactions. Biochim Biophys Acta 2000; 1477: 51-63.
[9] KITANO H, HIDAI C, KAWANA M and KOKUBUN S. An epidermal growth
42
factor-like repeat of Del1 protein increases the efficiency of gene transfer in vitro. Mol Biotechnol 2008; 39: 179-185.
[10] HIDAI C, ZUPANCIC T, PENTA K, MIKHAIL A, KAWANA M, et al. Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the alphavbeta3 integrin receptor. Genes Dev 1998; 12: 21-33.
[11] KITANO H, KOKUBUN S and HIDAI C. The extracellular matrix protein Del1 induces apoptosis via its epidermal growth factor motif. Biochem Biophys Res Commun 2010; 393: 757-761.
[12] KITANO H, MAMIYA A, KOKUBUN S and HIDAI C. Efficient nonviral gene therapy with FasL and Del1 fragments in mice. J Gene Med 2012; 14: 642-650.
[13] KITANO H, MAMIYA A, ISHIKAWA T, KOKUBUN S and HIDAI C.
Coagulation factor IX regulates cell migration and adhesion in vitro. Cell Biol Int 2015; 39: 1162-1172.
[14] LAWN R M. The molecular genetics of hemophilia: blood clotting factors VIII and IX. Cell 1985; 42: 405-406.
[15] LICHTMAN M A, ed., Williams HEMATOLOGY 7th, McGrawHill, New York, 2006.
[16] HEIT J A, THORLAND E C, KETTERLING R P, LIND T J, DANIELS T M, et al. Germline mutations in Peruvian patients with hemophilia B: pattern of mutation in AmerIndians is similar to the putative endogenous germline pattern.
Hum Mutat 1998; 11: 372-376.
[17] GREEN P M, BENTLEY D R, MIBASHAN R S, NILSSON I M and GIANNELLI F. Molecular pathology of haemophilia B. EMBO J 1989; 8:
43
1067-1072.
[18] HIRANO K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 2007; 27:
27-36.
[19] ZHAO P, METCALF M and BUNNETT N W. Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne) 2014; 5: 67.
[20] OWENS T W, VALENTIJN A J, UPTON J P, KEEBLE J, ZHANG L, et al.
Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ 2009; 16: 1551-1562.
[21] CHWA M, ATILANO S R, REDDY V, JORDAN N, KIM D W, et al. Increased stress-induced generation of reactive oxygen species and apoptosis in human keratoconus fibroblasts. Invest Ophthalmol Vis Sci 2006; 47: 1902-1910.
[22] FRISCH S M and RUOSLAHTI E. Integrins and anoikis. Curr Opin Cell Biol 1997; 9: 701-706.
[23] PAOLI P, GIANNONI E and CHIARUGI P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 2013; 1833: 3481-3498.
[24] BORUTAITE V, JEKABSONE A, MORKUNIENE R and BROWN G C.
Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol 2003; 35: 357-366.
[25] TAKEISHI A, KURANAGA E, TONOKI A, MISAKI K, YONEMURA S, et al.
Homeostatic epithelial renewal in the gut is required for dampening a fatal systemic wound response in Drosophila. Cell Rep 2013; 3: 919-930.
[26] ROSADO J A, LOPEZ J J, GOMEZ-ARTETA E, REDONDO P C, SALIDO G
44
M, et al. Early caspase-3 activation independent of apoptosis is required for cellular function. J Cell Physiol 2006; 209: 142-152.
[27] SHAW T J and MARTIN P. Wound repair at a glance. J Cell Sci 2009; 122:
3209-3213.
[28] HOFFMAN M, HARGER A, LENKOWSKI A, HEDNER U, ROBERTS H R, et al. Cutaneous wound healing is impaired in hemophilia B. Blood 2006; 108:
3053-3060.
[29] MONROE D M, MACKMAN N and HOFFMAN M. Wound healing in hemophilia B mice and low tissue factor mice. Thromb Res 2010; 125 Suppl 1:
S74-77.
[30] CHERA S, GHILA L, DOBRETZ K, WENGER Y, BAUER C, et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell 2009; 17: 279-289.
[31] GALLIOT B. Injury-induced asymmetric cell death as a driving force for head regeneration in Hydra. Dev Genes Evol 2013; 223: 39-52.
[32] TSENG A S, ADAMS D S, QIU D, KOUSTUBHAN P and LEVIN M.
Apoptosis is required during early stages of tail regeneration in Xenopus laevis.
Dev Biol 2007; 301: 62-69.
[33] FAN Y and BERGMANN A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell 2008; 14: 399-410.
[34] OKUYAMA R, NGUYEN B C, TALORA C, OGAWA E, TOMMASI DI VIGNANO A, et al. High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev Cell