ゲ ノ ム を 紡 ぐ
お願い および 注意事項
● 希望販売価格 ・・・「希望販売価格」は参考であり、販売店様からの販売価格ではございません。
記載の希望販売価格は 2015 年 1 月 1 日現在の希望販売価格です。
予告なしに改定される場合がありますので、ご注文の際にご確認ください。消費税は含まれておりません。
● 使 用 範 囲 ・・・記載の商品は全て、「研究用試薬」です。
人や動物の医療用・臨床診断用・食品用等としては使用しないよう、十分ご注意ください。
〒135-0016 東京都江東区東陽 2-2-20 東陽駅前ビル URL : http://www.cosmobio.co.jp/
取扱店
(12168)
1 コスモ・バイオのホームページへ Go!
このアイコンの数字が、
情報を得るための近道です !
お目当ての「記事 ID」を入力し、
検索をクリック!
3
www.cosmobio.co.jp
トップページの
「記事 ID 検索」を クリック!
2
まとめて検索 商品検索 カテゴリ検索 品番検索 SDS 検索 メーカー情報
検索 記事 ID 検索
0000
これだけ!
WEBへどうぞ
記事ID:●●●●●
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社
技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
広島大学大学院
理学研究科 数理分子生命理学専攻 教授
山本 卓
はじめに
ゲノム編集は、部位特異的ヌクレアーゼを用いて標的遺伝子を自 在に改変することが可能な新しい技術です。微生物から動物や植物 まで様々な生物での遺伝子改変が可能なことから、ゲノム編集はラ イフサイエンス研究において必要不可欠な技術となることが開発 当初から予想されていました。この予想は、第二世代の編集ツール TALEN と第三世代の編集ツールである CRISPR/Cas9 の開発に よって現実のものとなり、ゲノム編集技術は全ての研究者のための 技術となりつつあります。特に、第三世代の CRISPR/Cas9 は簡 便かつ効率的であることから、今後のゲノム編集研究は CRISPR/
Cas9 を中心として展開していくことは疑いのないところです。ゲ ノム編集のように開発スピードの速い技術は近年例がなく、基礎研 究のみならず応用研究での利用も積極的に進められています。本稿 では、ゲノム編集ツール開発の歴史、ゲノム編集を用いた標的遺伝子 改変、off-target 作用を抑えるゲノム編集技術の派生技術について 紹介します。
ゲノム編集ツール開発の背景
ゲノム編集技術の基盤となる部位特異的ヌクレアーゼは、人工ヌ クレアーゼと RNA 誘導型ヌクレアーゼに大別されます。人工ヌク レアーゼとして開発された第一世代のゲノム編集ツールは、Zinc- finger nuclease (ZFN) です。ZFN は、目的の配列に特異的に結 合する Zinc finger リピートに制限酵素 FokI のヌクレアーゼド メインを連結した人工ヌクレアーゼで(図1)、一組の ZFN を発 現させることによって目的の配列に DNA 二本鎖切断(DSB)を 誘導することができます。ZFN は 1996 年に報告されて以来、
ゲノム編集での多くの先駆的な研究に実績を有しています。しかし ながら、特異性の高い ZFN の作製が難しいことから、現在広く利 用されるに至っていません。これに対して、第二世代のゲノム編集 ツ ー ル で あ る TALEN (Transcription activator-like effector nuclease) は 作 製 が 容 易 で あ る こ と か ら、2010 年 以 降、様 々 な 作 製 方 法(Golden Gate 法)が 開 発 さ れ、TALEN に よ る 遺 伝 子 改 変 が 急 速 に 進 み ま し た(1)。TALEN は、植 物 病 原 細 菌 Xanthomonas 属の TALE タンパク質を DNA 結合ドメインに 利用した人工ヌクレアーゼで(図1)、ZFN と同様に1組のヌクレ アーゼによって DSB を導入します。TALE タンパク質は、TALE リピートと呼ばれる繰り返しモチーフを中心にもち、このリピート 数に応じた塩基配列数(~40bp)を認識することができるので、特 異性の高い TALEN の作製が可能です (TALEN では、~20bp を 利用している )。
RNA 誘導型ヌクレアーゼである CRISPR (Clustered regularly interspaced short palindromic repeats)/ Cas9(CRISPR- associated Protein9) は、2013 年の始めに彗星のごとく現れ ました(2)。人工ヌクレアーゼが、標的配列への結合に DNA 結 合ドメイン(タンパク質)を利用するのに対して、CRISPR/Cas9 は、RNA と標的 DNA の塩基対形成を利用します。細菌や古細菌 は、切 断 し た 外 来 DNA( フ ァー ジ な ど ) を CRISPR 領 域 に 取 り 込 み、こ れ を 鋳 型 と し た 短 鎖 の crRNA (CRISPR RNA) を 合 成 し ま す。さ ら に、crRNA と 別 の 短 鎖 RNA で あ る tracrRNA (transactivating CRISPR RNA) お よ び ヌ ク レ ア ー ゼ で あ る Cas9 の複合体が標的配列を切断します(Ⅱ型 CRISPR/Cas9)。
これによって、再び侵入してきた外来 DNA を不活性化すること ができるわけです。ゲノム編集では、crRNA と tracrRNA を1 分 子 の guide RNA(gRNA) と し て 作 製 し、gRNA と Cas9 の 2 種類の発現あるいは導入によって内在の遺伝子破壊が可能です
(図 1)。現 在 ほ と ん ど の 研 究 者 が、レ ン サ 球 菌(S. pyogenes)
の Cas9 を用いており、SpCas9 では標的配列の認識に PAM
(Protospacer Adjacent Motif)と呼ばれる配列が 3 塩基(NGG)
必要とされます。CRISPR/Cas の利点は、gRNA の種類を増やす ことによって、多重遺伝子改変が簡単に行えることで、培養細胞でも 生物個体でも多数の成功例が既に報告されています。
ゲノム編集を用いた標的遺伝子改変
ゲノム編集ツールによって導入された DSB は、細胞内の複数 の DNA 修復経路によって修復されます。主に、非相同末端連結
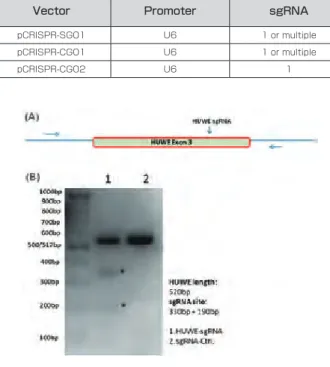
(NHEJ)と相同組換え修復(HR)の 2 つの経路によって修復さ れますが、繰り返し DSB を導入することによって修復エラーを誘 導し、標的遺伝子の改変を行います。NHEJ 経路では、修復過程の エラーによって、欠失や挿入などの変異を導入することが可能です。
これによって、遺伝子内の変異であれば、フレームシフトを引き起こ し遺伝子が破壊されます ( 図2 )。また、HR 経路の修復では、ド ナーベクターを共導入することによって、切断部分に外来の遺伝子 を挿入することが可能です ( 図2 )。最近では、ドナーベクターの 代わりに 1 本鎖 DNA を利用したノックインも盛んに行われてい ます。
NHEJ 経路を利用した遺伝子破壊は、基本的に全てのゲノム編集 ツールで可能であり、多くの成功例が報告されています(3)。最近 では、微生物などこれまで標的遺伝子改変が難しかった生物種での 改変例も増えてきています。一方、HR 経路を利用したノックイン は、細胞種や生物種によって効率が異なるので注意が必要です。HR の活性は、細胞周期に依存しており、全くノックインできないケース も見られます。また、ゲノム編集の効率は、細胞であれば遺伝子導入 効率、個体であればマイクロインジェクションが可能かどうかなど 実験手法によっても影響を受けます。
最近では、同時に複数箇所を切断することによって、染色体レベル での編集も可能であることが報告されています。同一染色体上の2 箇所を切断すると大規模な欠失を作製することが培養細胞や個体で 証明されています。欠失と比較すると頻度は低いですが、同一染色体 上の 2 箇所の切断によって逆位を誘導することも可能になってき ています。これらの染色レベルの実験を行う場合、CRISPR/Cas9 であれば複数のガイドを使うだけで実行可能です。筆者らは、最近 1 つのベクター中に 7 種類の gRNA の発現カセットを組み込ん だ all-in-one vector を開発しており、このベクターを用いること によって効率的に染色体レベルの改変が可能になると考えています。
ゲノム編集総説
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
ゲノムDNA ZFN
CRISPR/Cas9 Cas9
gRNA DNA結合ドメイン
人工ヌクレアーゼ
DNA切断ドメイン(Fokl)
{ {
RNA誘導型ヌクレアーゼ TALEN
| | | | | | | | | | | | | | | |
図 1. 各ゲノム編集ツールの概要
総 説
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社
技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Cas9 ニッカーゼと FokI-dCas9 を用いたゲノム編集 ゲノム編集技術では、類似配列への変異導入(off-target 作用)
について注意する必要があります。標的遺伝子の遺伝子破壊が成功 する一方で、類似する配列に欠失や挿入などの変異を同時に導入す る可能性です。off-target 作用の程度は、ゲノム編集ツールの種類 や用いる細胞や個体によって異なりますが、特にガン細胞では高い 傾向にあります(4)。
CRISPR/Cas9 においては、off-target 作用を低減させる目的 で、Cas9 の 2 つのヌクレアーゼドメインのうち 1 か所に変異 を入れた Cas9 ニッカーゼが開発されています(5)。近接する 2 箇所に gRNA を設計し、それぞれの DNA 鎖を Cas9 ニッカー ゼにより DNA ニックを入れると(Double nicking)、その領域で は DSB を導入された形になり、ゲノム編集が可能となります ( 図 3a)。gRNA が類似配列に結合した場合、DNA ニックが入ります が、欠失や挿入変異は導入されません。これによって、off-target 作用を低下させることができます。
さ ら に 最 近、高 活 性 か つ off-target 作 用 を 抑 え る CRISPR/
Cas9 として、Cas9 の 2 つのヌクレアーゼドメインの両方に変 異を入れた dCas (dead Cas9) を利用した方法が開発されまし た(6)。dCas9 の C 末端側に TALEN で利用されている制限 酵素 FokI のヌクレアーゼドメインを連結させ (FokI-dCas9)、近 接する 2 箇所に結合する gRNA と FokI-dCas9 によって標的 配列に DSB を導入できることが示されました ( 図 3)。この方法 は、1つの gRNA が類似配列に単独で結合しても DNA ニックや DSB を導入しないため、より安全な方法であると考えられます。
今後の展望
ゲノム編集は、CRISPR/Cas9 の開発によって全ての研究者の ための技術になったと言えます。研究者のアイディア次第でゲノム 編集を使った様々な研究が考えられます。最近、人工ヌクレアーゼ のヌクレアーゼドメインの代わりに転写因子の活性化ドメインや抑 制ドメイン、あるいは GFP を連結させた人工タンパク質の開発が 競って進められています。Cas9 についても dCas9 に様々な機
究にもゲノム編集を積極的に利用してほしいと考えています。
文献
1. Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39(12): e82, 2011 2. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu
PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems.
Science. 339(6121): 819-823, 2013
3. Peng Y, Clark KJ, Campbell JM, Panetta MR, Guo Y, Ekker SC. Making designer mutants in model organisms.
Development. 141(21): 4042-4054, 2014
4. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 31(9): 822-826, 2013
5. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 154(6):
1380-1389, 2013
6. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol.
32(6): 577-582, 2014
山本 卓(やまもと たかし)
広島大学大学院 理学研究科数理分子生命理学専攻 分子遺伝学研究室 教授
●経歴
1991 年 広島大学大学院理学研究科修了 1992 年 熊本大学理学部助手
1996 年 広島大学博士(理学)
2002 年 広島大学大学院理学研究科講師 2003 年 広島大学大学院理学研究科助教授 2004 年より現職
2014 年 鳥取大学と熊本大学の客員教授、
広島大学研究拠点「ゲノム編集研究拠点」リーダー
●主な専門・研究分野
ゲノム編集、発生生物学、ゲノム生物学
●学会等活動
ゲノム編集コンソーシアム(代表)
●最近の代表的論文
1. Sakuma T, Nishikawa A, Kume S, Chayama K and Yamamoto T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system.
Scientific Reports, 4: 5400, 2014
2. Sakuma T, Hosoi S, Woltjen K, Suzuki KI, Kashiwagi K, Wada H, Ochiai H, Miyamoto T, Kawai N, Sasakura Y, Matsuura S, Okada Y, Kawahara A, Hayashi S and Yamamoto T. Efficient TALEN construction and evaluation methods for human cell and animal applications. Genes Cells, 18: 315-326, 2013
3. Ochiai H, Sakamoto N, Fujita K, Nishikawa M, Suzuki KI, Matsuura S, Miyamoto T, Sakuma T, Shibata T and Yamamoto T. Zinc-finger nuclease-mediated targeted insertion of reporter genes for quantitative imaging of gene expression in sea urchin embryos. Proc Natl Acad Sci U S A, 109: 10915-10920, 2012
著者プロフィール
Cas9 (D10A)
dCas9 (D10A, H840A) Cas nikase
Folk-dCas9 gRNA1
gRNA1
gRNA2
gRNA2 DNA 切断とメイン (Fok1)
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | || | | | | | | | | | |
| | | | | | | | | | | | | | | || | | | | | | | | | |
挿入・欠失による 遺伝子ノックアウト
× ×
遺伝子ノックイン GFP
GFP HR による 外来遺伝子の挿入 NHEJ の修復エラー
部位特異的ヌクレアーゼによる DSB の誘導
図 2. 部位特異的ヌクレアーゼによるゲノム編集概略
図 3. ダブルニッキング法と FokI-dCas9 法の概略
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社
技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
CRISPR-Cas(clustered regularly interspaced short palindromic repeats-CRISPR associated proteins)シ ス テ ム は、細菌や古細菌においてウイルスやプラスミドといった遺伝的要 素の侵入物を標的し、排除するよう進化した適応免疫の一つである。
これらのシステムは、cas 遺伝子カセットと外来性 DNA に由来 して、遺伝記憶となる短鎖と特徴的な「スペーサー」配列を間に挟 んだ一連の直列反復配列をコードする CRISPR アレイから構成さ れる。転写および CRISPR 座位の成熟を経て、Cas タンパク質と ともに外来性 DNA 検出の監視システムとして機能する CRISPR RNAs(crRNAs)が産生され、やがて RNA にガイドされた干渉 により外来性 DNA を崩壊へと導く。細菌性Ⅱ型 CRISPR-Cas シ ステム由来のヌクレアーゼ Cas9 は形質転換された生物工学ツー ルであり、現在ではシンプルなワトソン - クリック塩基対を基にし たゲノム編集の重要なツールとして利用されている。簡便かつ効率 がよいことから、実質的に全てのゲノムを標的することができ、世界 中の数々の研究室が急速に採用している。さらに、本システムはゲノ ム編集のみに留まらず、遺伝子発現のオン / オフや生細胞における 画像診断においても利用されている。
CRISPR/Cas システムの基礎
2007 年に、CRISPR/Cas システムが適応免疫に関与するこ とが始めて実験的に示された。ストレプトコッカス・サーモフィル スに溶菌性ファージを感染させると、その後ファージに対する免疫 が得られることが報告されている。その翌年、 成熟した crRNA が Cas タンパク質との複合体内でガイドとして機能し、細菌における ウイルス DNA 増殖と干渉を誘導することが発見された。現在では 3段階で適応免疫が生ずることが知られている。
ステップ①: 侵入 DNA の短鎖配列獲得(プロトスペーサー)およ びスペーサーとして CRISPR アレイへの挿入。これ には、Cas タンパク質によるプロトスペーサー切断お よび CRISPR アレイでの反復複製とその後の組み換 えが含まれる。
ステップ②:pre-crRNA の転写が生じた後、成熟 crRNA 産生プ ロセスを経由する。何れも反復配列と侵入物標的ス ペーサーをもつ。
ステップ③:crRNA スペーサー配列相補部位において Cas タン パク質による外来性核酸の crRNA 先導型切断が生ず る(干渉)。
3種(Ⅰ型、Ⅱ型、Ⅲ型)の CRISPR-Cas システムが存在し、干 渉の際、各々異なる分子機序を利用する。Ⅰ型とⅢ型システムでは crRNA 先導型ターゲティングにおいて Cas タンパク質の大型複 合体を利用するが、Ⅱ型システムでは RNA 先導型 DNA 認識と切 断に Cas9 タンパク質のみを必要とする。このⅡ型の特性はゲノム 工学アプリケーションにおいて非常に有用であることが立証されて いる。
Cas9-sgRNA 複合体の構造および機能
初期の遺伝学研究により、細菌において Cas9 がウイルス防 御 に 必 須 で あ り、侵 入 DNA に 二 本 鎖 切 断 (DSBs) を 導 入 し、
in vivo DNA 標的を可能とすることが示されていた。2012 年
に、Cas9 が tracrRNA と crRNA の2種 の RNA を 利 用 し て DNA 切断を誘導することが示された。標的認識には crRNA 配 列との塩基対形成と標的配列隣接部位の PAM の存在が必要であ
CRISPR/Cas システム
非常に優れたゲノム編集システム
る。tracrRNA-crRNA 複合体は sgRNA として設計することが できる。5' 末端の 20 塩基配列が DNA 標的部位をワトソン-
クリック塩基対形成により決定し、3' 末端の二本鎖ループ構造が Cas9 に結合するという2つの重要な特色をもつ。本発見により、
sgRNA 内の 20 塩基のガイド配列を変更することで、PAM 配列 に隣接した如何なる DNA 配列をも標的できるシステムが構築され た。TALEN や ZFN のような、各 DNA 標的部位に対応したタ ンパク質エンジニアリングが必要となるゲノム編集手法と異なり、
CRISPR-Cas9 システムではガイド RNA 配列の変更のみで標的 ゲノムおよびそれに対する特異性を変更することができる。
概 論
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社
技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説 CRISPR/Cas9 システムは、カスタム化(標的遺伝子の変更や
複数遺伝子のターゲット)が容易であることから、現在、細菌、寄 生生物、ゼブラフィッシュ、マウスおよびヒト細胞をはじめとした 膨大な種類の細胞や生物種において、そのゲノム編集または修正に 利用されている。カスタムデザインされた sgRNAs と共にヒト細 胞(胚胎性腎臓細胞、慢性骨髄性白血病細胞、iPS 細胞など)内で 発現させると Cas9 はゲノム DNA 内に DSBs を産生し、その 後、NHEJ( 非相同末端結合 ) により修復され遺伝子破損を生ずる か、HDR( 相同組換え修復 ) によりドナー遺伝的配列の挿入が生 ずる。決定された部位に DSBs を導入することで、肺がん、急性 骨髄性白血病およびユーイング肉腫において生ずるものと類似した 染色体転座をもつヒト細胞株および初代培養細胞が産生される。さ らに、multiplexing として知られる複数の sgRNA を用いた複 数座位の同時標的も可能であることが報告されている。CRISPR- Cas9 は既にβ型地中海貧血症、チロシン血症および嚢胞性線維症 といった特定遺伝子内の変異修復にも使用されている。したがって、
CRISPR-Cas9 は、ゲノム再編成研究や癌などのの疾患発生のさら なる理解においてロバストかつ順応性のあるツールとなり得ると同 時に遺伝学を基盤とした治療の可能性を秘めている。
その他の応用
・生細胞のイメージング
CRISPR/Cas システムでは、gRNA により如何なる配列も標 的・編集することができるため、生細胞における特定染色体部位の イメージングへの応用が期待できる。ヌクレアーゼドメインが非活 性化された修正型 Cas9 タンパク質(dCas9)を蛍光タンパク質
(EGFP など)に融合させ、特異的にデザインされた sgRNA とと もに用いることで、dCas9 は生細胞における翻訳領域および非翻 訳領域の DNA 画像化に利用することができる。このようなイメー ジングツールは、生細胞における天然の染色体の高次構造動態研究 用に用いられている現在の技術改良に実質上利用できる可能性があ る。蛍光タンパク質または小分子を sgRNA にカップリングできる 可能性もあり、Cas9 を利用した多色画像化に新たな方略を与える ことが考えられる。
・遺伝子制御
Cas9 の主な特徴はガイド RNA 配列と PAM により決定さ れた DNA 部位に結合できることであり、DNA の永久的な改変を 超えた応用が可能となる。特に、dCas9 と設計された sgRNA の 組み合わせにより、ゲノムワイドスケールでの標的遺伝子制御へ と目的が移行している。CRISPRi として知られるプロセスでは、
RNAP の DNA アクセスを dCas9 により遮断することもできる ため、細菌とヒト細胞において転写を可逆的に抑止できる。さらに、
RNAP ω - サブユニットなどの調節ドメインと融合させたキメラ dCas9 を構築することで、CRISPRi を効果的な遺伝子活性化へと 利用できる可能性も考慮されている。
参考文献
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007)
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012)
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2, e00471, (2013)
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013) Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013)
Sternberg, S. H. et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014)
Jinek, M. et al. Structures of Cas9 endonucleases reveal RNA mediated conformational activation.
Science http://dx.doi.org/10.1126/science.1247997 (2014)
van der Oost, J. et al. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nature Rev. Microbiol. 12, 479–492 (2014)
Anders, C. et al. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease.
Nature http://dx.doi.org/10.1038/ nature13579 (2014)
Cosmo Bio would like to acknowledge and thank the OriGene Technologies, Inc. for providing CRISPR/Cas system information presented here.
Acknowledgements
This airticle was put together with scientific input from Jennifer A.Doudna, Tom Riis and Sam
Sternberg, Department of Molecular and Cell Biology and Department of Chemistry,Howard Hughes Medical Institute, University of California Berkeley,Berkeley, California 94705, USA.
The authors have no affiliation with OriGene.
© 2014 Nature Publishing Group. All rights reserved.
http://www.nature.com/nrmicro/posters/crispr/ index.htm
Applied Biological Materials 社
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用ガイド RNA ライブラリー 低コスト! CRISPR/Cas9 技術を用いたゲノムワイド sgRNA コレクション
APB
WEBへどうぞ
記事ID:13443
Applied Biological Materials 社
iCRISPR/Cas9 ゲノム編集用ガイド RNA ライブラリー
ゲノム編集において、RNA 先導型の最新のエンドヌクレアー ゼ ツ ー ル に CRISPR /Cas9 シ ス テ ム (Clustered Regularly Interspaced Short Palindromic Repeats:規則的な間隔をもっ てクラスター化された短鎖反復回文配列 ) があります。本システム では、単鎖ガイド RNA(sgRNA)を使用して特異性の高いゲノム 破壊や置換を行います。この簡便かつロバストなゲノム編集システ ムは真核生物細胞に利用可能です。相同組換えにより対象ゲノムを 希望する部位で切断することで、非常に簡便に、自由に、かつ効果的 に遺伝子発現抑制や遺伝子編集を実現します。
ウイルスベクターや遺伝子工学の分野で長年実績のある Applied Biological Materials 社 で は、こ の 最 新 の 技 術 を 利 用 す る た め の製品を低価格でご提供しています。非組込み型アデノウイルス sgRNA 発現システムをはじめ、全ヒト、マウス、およびラット遺 伝子を標的とした独自のゲノムワイド sgRNA ライブラリー発現 用のレンチウイルスシステムを開発しました。レンチウイルスシ ステムは、実験手技等に左右されず、簡便で、Applied Biological Materials 社独自の Cas9 ヌクレアーゼやニッカーゼ発現ベク ターおよびウイルスと補完することで、iCRISPR システムは遺伝 子ノックアウト実験において理想的な手法となります。
Cas9 発現
○ レンチウイルスベクター
○ パッケージ済みレンチウイルス
○ パッケージ済みアデノウイルス
ゲノムワイド iCRISPR sgRNA ライブラリー
● 野生型 Cas9 ヌクレアーゼ
● 改変型 Cas9 ニッカーゼ
○ レンチウイルスベクター
○ パッケージ済みレンチウイルス
○ パッケージ済みアデノウイルス
○ ヒト、マウス、ラット 受託サービス
● ノックアウト細胞株構築
● カスタム標的レンチウイルス sgRNA ライブラリー
iCRISPR システムは、特異性の高いゲノム崩壊と置換が可能で す。この簡便かつロバストなシステムでは、
(1) Cas9 ヌクレアーゼまたは Cas9 ニッカーゼ (2) 標的特異的単鎖ガイド RNA(sgRNA)
の 2 つの要素が必要です。
レンチウイルスベクターの形質転換、レンチウイルスのトランスダ クション、またはアデノウイルスのトランスダクションにより標的 細胞や宿主における Cas9 発現が生じます。sgRNA がゲノム上 の標的配列をみつけると、Cas9 ヌクレアーゼがゲノム DNA の両 鎖を切断し二本鎖切断(DSBs)が生じます。これが修復されると 挿入欠失フレームシフトまたは早期の停止コドンが生じます。
自由度が高く多様性のあるゲノム編集ツール“ iCRISPR システム”
二本鎖切断(DSBs)
挿入欠失フレームシフト 早期の停止コドン
非相同末端結合による修復
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用ガイド RNA ライブラリー
Cas9の種類 製品種 メーカー略号 品番 希望販売価格
ヌクレアーゼ(野生型)
レンチウイルスベクター APB K002 ¥69,000
レンチウイルス APB K003 ¥111,000
アデノウイルス APB K004 ¥111,000
ニッカーゼ(改変型)
レンチウイルスベクター APB K005 ¥69,000
レンチウイルス APB K006 ¥111,000
アデノウイルス APB K007 ¥111,000
Guide RNA 要件 インデルフレーム
シフト誘導 HDR 誘導 推奨アプリケーション
Cas9ヌクレアーゼ 各標的遺伝子につき 1つのgRNA
Yes
最も効果的 Yes InDel, HDR (正確さが重要で、オフターゲット作用が それほど問題でない場合)
Cas9 ニッカーゼ
各標的遺伝子につき
1つのgRNA 少々
Yes
あまり効果的で ない
HDR経路のみ、InDel生成を必要としない場合 Cas9
ニッカーゼ
各標的遺伝子につき 2つのgRNA
Yes
非常に効果的 Yes InDel, HDR (正確さが重要、かつオフターゲット作用が 問題視される場合)
細胞は、非相同末端結合(NHEJ)経路のほかに相同組換え修 復(HDR)により DNA 修復を行うことができ、この場合、ゲノ ム DNA に核酸変異の導入を行うことがあります。目的配列をもつ DNA 修復の鋳型は、通常、sgRNA/Cas9 とともに細胞に形質転 換しますが、二本鎖切断に近接の上流または下流の配列と高度に相 同性をもつ必要があります。Cas9 ヌクレアーゼにより導入された 二本鎖切断は、HDR を受けた後、特定変化がゲノム DNA 内に永 久に導入されます。これは、改変型 Cas9 ニッカーゼにより導入さ れた一本鎖切断でも同様です。
Cas9 ヌクレアーゼの触媒領域の1つを不活性化させた「ニッ カーゼ」は、ゲノム DNA 上の標的部位における二本鎖切断ではな く一本鎖切断を行います。期待する二本鎖切断を行うため、1つでは なく2つの gRNAs をゲノム DNA の近接領域(20 塩基未満)
の両鎖に配置した場合、ガイド RNA(gRNA)がミスマッチをもっ て期待しない DNA 部位に結合しオフターゲット作用を生ずること がありますが、改変したニッカーゼによりをこの現象を削減するこ とができます。ゲノム DNA 上の期待しない単鎖の切れ目は、無傷 の反対鎖を鋳型として相同配向性修復(HDR)経路により直ちに修 復されます。
Cas9 ヌクレアーゼによる相同組換え修復(HDR)
特異性と精度を改変した Cas9 ニッカーゼ
Applied Biological Materials 社
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用ガイド RNA ライブラリー
1 コスモ・バイオ WEB ページの詳細検索をクリック。
2 キーワード検索にご希望の遺伝子名、品番に「K* ( アスタリスク )」、
メーカー略号に「APB」を入力して検索してください。
例:DUX4L7
3 検索結果が表示されます。
品番、品名、数量を指定し、ご利用の代理店様へご注文ください。
CRISPR/Cas9 システムを利用することで、非常に特異的な ゲ ノ ム 編 集 が 可 能 に な り ま す。標 的 特 異 的 な 単 鎖 ガ イ ド RNA
(sgRNA)に先導され、Cas9 ヌクレアーゼがゲノム DNA の二 本鎖を巻き戻し sgRNA による標的配列を認識して両鎖を切断し ます。これにより生ずる二重鎖切断は非相同末端結合(NHEJ)経 路により修復され、標的遺伝子の翻訳領域が崩壊します。Applied Biological Materials 社は、レンチウイルスベクターやすぐに実 験にご便用頂けるレンチウイルスおよびアデノウイルスを利用し た、ヒト、マウス、およびラット遺伝子を標的とするゲノムワイド iCRISPR sgRNA ライブラリーを開発しました。
Applied Biological Materials 社のレンチウイルス iCRISPR sgRNA ベクターおよびウイルスは、1種類ずつ、または 3 種の セットでご提供していますので、実験条件にあわせてお選びくださ い。個別使用または複数をプールして使用するなど、実験系に適した 遺伝子ノックアウト実験を行って頂けます。
iCRISPR sgRNA アデノウイルスは、特に導入困難な細胞種を始 めとした幅広い標的細胞において、非組込み型かつ高い効率でのゲ ノム編集に適しています。
ゲノムワイド sgRNA(単鎖ガイド RNA)コレクション
■ 野生型 Cas9 ヌクレアーゼ用 sgRNA コレクション
品名 製品種 価格
各 sgRNA
レンチウイルスベクター ¥38,000
レンチウイルス ¥119,000
アデノウイルス ¥251,000
3 種の sgRNA セット レンチウイルスベクター ¥105,000
レンチウイルス ¥279,000
■ ニッカーゼ(改変型 Cas9)用 sgRNA コレクション
品名 製品種 価格
各 sgRNA
レンチウイルスベクター ¥76,000
レンチウイルス ¥238,000
アデノウイルス ご照会
3 種の sgRNA セット レンチウイルスベクター ¥210,000
レンチウイルス ご照会
Applied Biological Materials社より ご提供
パッケージ済み
レンチウイルス Cas9とsgRNAウイルス
を同時導入した標的細胞 遺伝子発現レベル 変化
GeneCopoeia 社 Santa Cruz Biotechnology 社 技術情報 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR カスタムノックアウトサービス
【 お問い合わせ先 】
ご質問・ご不明の点は技術サービス部 テクニカルサービスグループまでお問い合せください。
また、秘密保持契約のご希望につきましても、下記までご連絡をお願いいたします。
TEL : 03-5632-9615 FAX : 03-5632-9614
E-mail : [email protected]
【 お問い合わせ先 】
ご質問・ご不明の点は技術サービス部 テクニカルサービスグループまでお問い合せください。
また、秘密保持契約のご希望につきましても、下記までご連絡をお願いいたします。
TEL : 03-5632-9615 FAX : 03-5632-9614
E-mail : [email protected] APB
WEBへどうぞ
記事ID:13443
Applied Biological Materials 社
iCRISPR プール型カスタム sgRNA レンチウイルスライブラリー作製サービス
APB
WEBへどうぞ
記事ID:13443
Applied Biological Materials 社
iCRISPR カスタムノックアウト細胞作製サービス
プール型カスタム sgRNA レンチウイルスライブラリーを利用 することで最大 100 種の標的遺伝子を、一度にノックアウトする ことができます。本商品はお客様のご研究に合わせて作製しますの で、特に遺伝子ファミリーや経路のノックアウトに有用です。プール 型レンチウイルス粒子をご提供致します。ご希望の遺伝子の生物種、
遺伝子名およびアクセッション番号を表記した標的遺伝子リストを コスモ・バイオへご連絡ください。
本サービスでは、如何なる細胞株のどの遺伝子であってもノック アウト致します。Applied Biological Materials 社へご希望の標 的細胞と、ノックアウトしたい遺伝子の生物種、遺伝子名およびアク セッション番号をご連絡頂くだけです。ゲノム編集に成功した細胞 は、厳格な品質管理と遺伝子ノックアウトの検証後、お客様にお届け 致します。
sgRNA レンチウイルス
プール型 sgRNA レンチ ウイルスライブラリー
sgRNA プールと Cas9 レンチウイルスを標的細胞 に同時に導入
二本鎖切断
NHEJ による修復後、
挿入欠失変異
Cas9 guide
RNA
sgRNA および Cas9 ウイルスを 標的細胞に導入
標的遺伝子 ノックアウト
ワイルドタイプ ノックアウト
GeneCopoeia 社
Applied Biological Materials 社
Santa Cruz Biotechnology 社 技術情報 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用製品
■ 技術情報
Q
【01】Applied Biological Materials 社で保証できるノックアウト効率を教えてください。また、3 種の gRNA の何れも標的をノックアウトできなかった場合、他の候補を提供してもらえますか?
A
Applied Biological Materials 社の経験では、デザインしたコンストラクトの 90% 以上が 20 ~ 90% の細胞において完全な ノックアウトを達成しています。感染後ピューロマイシン選択した細胞の 50% 以上において、ほとんどのコンストラクトから完全な ノックアウトが得られています。実証例から sgRNA は shRNA や siRNA よりかなり優秀であることが示唆されます。3 コンスト ラクトのいずれからも顕著なノックアウトが得られないというのは非常にまれな状況です。もし、いずれのコンストラクトを用いても適切なノックアウトが得られない場合、異なる 3 種のコンストラクトを無料でデザイン致し ます。交換は1回のみに限ります。交換には細胞のモニターが必須条件で、Surveyor アッセイの分析証明をご提供ください。標的配列 増幅用 PCR プライマーを設計し T7E で分解させます。
Q
【02】本システムを長鎖ノンコーディング RNA に使用した経験はありますか。A
本システムでは、長鎖ノンコーディング RNA や miRNA、もしくは短鎖 DNA 断片を二本鎖 sgRNAs によりノックアウトすること も可能です。しかし、ノックアウト効率はタンパク質コーディング遺伝子に比べて非常に低く、~ 1% 程度です。したがって、単一細胞 クローニングが必要となります。Applied Biological Materials 社では、lncRNA の最初と最後を標的する 3 種の sgRNAs をデ ザインし、今後上市する計画もあります(3x2 コンストラクト)。標的 lncRNA のノックアウト用に 3x3 の組み合わせがあるため、PCR スクリーニングにより最良の組み合わせを確認します。その後、2つの最もよい sgRNAs をトランスダクションした後、単一細 胞クローニングします。1 ~ 5% のクローンにおいて lncRNA の完全ノックアウトが期待されます。
Q
【03】ノックアウトがみられるまでにどのくらいの時間が必要ですか。A
ピューロマイシン選択後1週間です。Q
【04】推奨する培養条件を教えてください。A
レンチウイルストランスダクションの標準型プロトコールに準ずるとともに、ご利用の特定細胞に対して最適化された培養条件をお確 かめの上ご利用ください。Q
【05】sgRNA の代替品を要求する場合のガイドラインを教えてください。A
Applied Biological Materials 社では、セットでご購入いただいた 3 種の sgRNA レンチウイルスコンストラクトのうち少なくと も1つは、トランスダクションと薬物選択後、50% 以上の細胞においてフレームシフト変異による完全な遺伝子ノックアウトを示すこ とを保証します。遺伝子ノックアウト効率は Surveyor アッセイで素早く検査でき、ウェスタンブロットまたは他の機能解析により確 かめることができます。非常にまれな事例ではありますが、もし顕著な効果が得られない場合は、異なる sgRNA 配列をもつ 3 種の新 規コンストラクトを代替品として1回のみご提供致します。代替品提供をご希望の場合、まず Surveyor アッセイ結果をご提示頂いた 上で Applied Biological Materials 社にて内容を確認致します。代替品セットは保証対象外です。もしこれらのコンストラクトも効果 がないと考えられる場合、おそらく理由は原因不明であると思われます。Applied Biological Materials 社では、本技術の成功に向けてその義務と責務を制限するため、如何なる sgRNA レンチウイルス製 品においても代替品のご提供は1回のみとしています。お問い合わせ頂く前に、お客様において MOI の増大(10 程度まで)、感染時 間(最長 72 時間)およびノックアウトアッセイの前にクローンのスクリーニングを行うなど、可能な限り実験系の最適化を行ってい ただけますようお願い致します。
iCRISPR/Cas9 ゲノム編集用製品 FAQ
GeneCopoeia 社 Santa Cruz Biotechnology 社 技術情報 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用製品
iCRISPR/Cas9 ゲノム編集用 iCRISPR 製品 プロトコール
Applied Biological Materials 社では、プラスミド DNA を 10mM Tris に入れてご提供しており(特別なご指定のない限り)、標準的な DH5 αE.coli 株に直接形質転換することを想定しています。Applied Biological Materials 社より入手した何れのプラスミドを増幅す る場合も、DH5 αコンピテントセルに形質転換し、単一コロニーを拾って一般的なプラスミドミニプレップを行います(タンパク質発現用
E.coli 株はプラスミド抽出や精製に向かない場合があるため、プラスミド増幅への使用はお奨めしません。)。プラスミドの抗生物質選択を確
認してください。コードされる耐性遺伝子は通常、アンピシリン、カナマイシンまたはスペクチノマイシンですが、テトラサイクリンやクロラ ムフェニコールをもつ場合もあります。Applied Biological Materials 社の pLenti ベクターは全て高コピー数プラスミドです。
■
E.coli
DH5 αコンピテントセルのサブクローニング効率● 最良の結果を得るために Applied Biological Materials 社の ProClone™ Competent Cells (品番: E003)をご利用ください
● コンピテントセルは 4x1.25mL 容量で提供されます。-80℃で保存してください。
● 凍結溶解の繰り返しを避けるため、分注保存してください。
● 湿った氷上にバイアルを静置して溶解します。細胞を分注する前に新しいチューブを冷却しておきます。
● 溶解後、ゆっくりと反転させて細胞を混合します。
● 直ちに 50 μ L ずつ分注し、ドライアイス/ 95% エタノール内で再凍結します。
■ 形質転換プロトコール
1. ProClone™ Competent DH5 α cell を湿った氷上で溶解する。
2. 1 μ L のプラスミドを添加する。プラスミド濃度が既知の場合、10ng/ μ L に希釈して 1 μ L 使用する。
チューブを軽くたたいて混合する。氷上で 30 分間静置する。
3. 42℃のウォーターバス内で 45 秒間きっちりと熱処理する。
4. チューブを氷上に戻し2分間静置する。
5. 150 μ L の滅菌済み LB 培地を加え、37℃、240rpm に設定した培養振とう器内で1時間振とう培養する。
6. 全量を適切な抗生物質を含む LB 寒天培地上に撒く(濃度は下記参照)。
7. 37℃で一晩培養(16 時間程度)してコロニーを形成させる。もしコロニーが凝集している場合は、1 μ L のセルを 100 μ L の LB に加え、新しい抗生物質入り LB プレートにまく。
8. 4 ~ 10mL の抗生物質を含む LB 培地に単一コロニーを接種する。37℃、240rpm に設定した震盪培養器で一晩(16 ~ 18 時間)
培養する。
9. 一般的なミニプレッププロトコールに準じてプラスミドを単離する。
抗生物質選択
KanR: 50 μ g/ml カナマイシン
AmpR: 100 μ g/ml カルベニシリン/アンピシリン SpecR: 50 μ g/ml スペクチノマイシン
TetR: 12.5 μ g/ml テトラサイクリン
CamR: 25 to 34 μ g/ml クロラムフェニコール
プラスミド増幅
以下は、最大 106 IU/mL タイターの組換え型レンチウイルス粒子産生用プロトコールです。実験結果の評価の手だてとするため、ネガティ ブコントロール(DNA またはトランスフェクション試薬なし)のご使用をお奨めします。
レンチウイルスパッケージングを始める前に、適切な量の発現 DNA(10 μ g プラスミド/ 10 cm ディッシュ)があることを確認し ます。DNA 増幅ステップでは、通常、標準的な細菌の形質転換プロトコールを利用します。Applied Biological Materials 社では、E.coli DH5 α株によりレンチウイルスパッケージング用プラスミドが高収量で産生でき、また組換えが生ずるリスクが低いことを確認しているた め、ご提供した DNA プラスミドの増幅にはE.coli DH5 α株のご利用をお奨めします。
レンチウイルスパッケージング
GeneCopoeia 社
■ 技術情報
Applied Biological Materials 社
Santa Cruz Biotechnology 社 技術情報 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用製品
重要な注意事項
哺乳動物細胞へのトランスダクション効率は実験条件下によって顕著に異なります。感染に使用したウイルス濃度、ウイルスへの曝露 時間、およびウェルやプレートの生長領域なども関与します。対象とする標的細胞における望ましい感染効率(MOI)を知るのに必要な ウイルス濃度を決定するためには、レポーター遺伝子をもつウイルス粒子(例:GFP コントロール 品番:LV006 またはβ -gal コン トロールレンチウイルス 品番:LV007)を利用したトランスダクションの予備試験を 1 μ l, 5 μ l, 10 μ l および 100 μ l. と いった異なる容量範囲において複数回行うことが理想的です。この予備試験結果を最も感染細胞の割合が高くなると思われる至適濃度決 定に利用します。
抗生物質選択をしない場合、下流のアッセイはトランスダクションから 48 ~ 72 時間後に行います。抗生物質選択を行うか否か は標的細胞のトランスダクション効率や増殖率ならびに計画している生物学的アッセイに応じて決定します。感染効率の高い細胞(例:
HEK293, HT1080, HeLa, MDA-MB0468 細胞など)の場合、抗生物質選択を行うことなくほとんどの生物学的アッセイを行うこと ができます。感染に耐性の細胞に対しては、レンチウイルスコンストラクトを提要発現しているクローンのみを選択して下流のアッセイ を行うことが理想的です。
1日目 1. 午後に~ 1.2 x 107 293T 細胞を 10cm ディッシュに播種する。
2日目
(ステップ2~6を トランスフェクション当日の
午前中に行う)
2. 細胞密度が70~80%であることを確認する。
3.
a) 10 cmディッシュごとに下記に準じてトランスフェクション複合溶液を調製する。
溶液A: 20 μgのDNAプラスミド(発現ベクター10μgとApplied Biological Materials社の第2世代
(品番:LV003)または第3世代(品番:LV053)パッケージングミックス 10μg)を1mLの無血 清かつ抗生物質を含まない培地に加えて希釈する。
溶液B: 80μLの LentiFectin™ トランスフェクション試薬(品番:G074)を1mLの無血清かつ抗生物 質を含まない培地に添加する。
b) 上記溶液を室温で5分間静置する。
c) 溶液Aと溶液Bをよく混合し、室温で20分間静置する。これによりトランスフェクション複合体が形成さ れる。
4. 4.5mLの無血清培地をトランスフェクション複合体に添加する。
5. 10cmディッシュに播種している細胞より培地を除去する。
6. ステップ4で調製したトランスフェクション複合体を細胞に添加し、37℃で5~8時間培養する。細胞が外れ ないようディッシュの壁面よりゆっくりと混合溶液を添加する。
7. 10cmディッシュに0.65mLのFBSを添加し、37℃で一晩培養する。
3日目
8. 細胞よりトランスフェクション培地を除去する。
9. 10mL の完全培養培地を細胞に添加する。
10. 37℃で 24 時間培養する。
4日目(回収)
11. 培養ディッシュより上清培地を回収する。
12. 上清を 4℃、3000rpm で 15 分間遠心し、細胞の残渣を沈降させる。
13. 清澄な上清を新しいチューブに移しとり、低タンパク質結合性の 0.45 μ M 滅菌フィルターでろ過する。
14. 最初に回収したウイルスタイターは 106 IU/mL 程度である。ろ過した上清はin vitro 感染や濃縮に使用 できる。または、-80℃で保存することもできる。凍結溶解の繰り返しによるウイルスタイターの低下を避 けるため、長期保存の場合は分注することが望ましい。
15. 1 回目の回収後、細胞に 10mL の完全培地を加えて 37℃でさらに 24 時間培養し、2 回目の回収を行 うこともできる。1回目のウイルス上清を 4℃で一晩保存し、翌日に2回目のウイルス上清をこれに追加す ることもできる(上清を凍結するとタイターの顕著な低下を招く恐れがある)。
5日目
16. ステップ 11 ~ 13 に準じて 2 回目の上清回収を行い、1 回目のウイルス上清に加える。
17. ウイルスタイターが 106 IU/mL より高い場合、Applied Biological Materials 社の qPCR レンチウイ ルスタイターキット(品番:LV900)を用いて迅速かつ簡単にタイター測定が可能。濃縮が必要な場合に は、Applied Biological Materials 社の Ultra Pure Lentiviral Purification Kit(品番:LV998)によ りウイルス濃縮をすることも可能。
標的細胞へのレンチウイルス感染
■ レンチウイルスパッケージングプロトコール
注: VSV-G 糖タンパク質発現は 293T 細胞の融合の原因となるため、融合細胞として知 られる大型の複数核細胞様式を示す。この形態変化は正常であり、レンチウイルス産生 には影響しない。
GeneCopoeia 社 Santa Cruz Biotechnology 社 技術情報 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
Applied Biological Materials 社 iCRISPR/Cas9 ゲノム編集用製品
■ 感染プロトコール
下記のプロトコールは一般的なガイドラインとしてご提供していますので、標的細胞へのトランスダクションにおける最適条件を決定するた めの出発点としてご利用ください。
1. ウイルス感染の 24 時間前に標的細胞を 24 穴プレートの各ウェルに 0.5 × 105 細胞ずつ播種する。0.5mL の完全至適培地(血清 と、必要であれば抗生物質を含む)を添加後、5% CO2 存在下の 37℃で一晩培養する。
2. ポリブレンを完全培地に加えて 8 μ g/mL 濃度とする。ウェルより生育培地を除去し、ウェルごとに 0.5mL のポリブレン/培地混合 溶液を添加する(培地容量は使用するプレートサイズに準じて適宜変更する)。標的細胞の感染効率が低い場合、ViralPlus Transduction Enhancer ( 品番:G698) を 1:100(または最適化させた希釈率)で添加する。
3. 予備実験より、標的細胞に対する有効な MOI が決定できている場合、これに準じて適切なウイルス量を標的細胞に感染させる。Cas9 と sgRNA レンチウイルスは標的細胞に対して同時感染することもできる。
ポジティブコントロールとして使用するウェルの1つに GFP コントロールウイルスを使用するとともに、他のウェルの1つに空のウイ ルスコンストラクトをコントロールとして使用することをお奨めします。1つのウェルはウイルスを感染せずに放置して標準コントロール とする。
4. 感染後、細胞を 5% CO2 存在下の 37℃で一晩培養する。
5. 培養溶液を除去し、1mL の完全培地と置換する。細胞は 5% CO2 存在下の 37℃で一晩培養する。
6. 翌日、細胞を 1:3 または 1:5 に分割(使用細胞の細胞成長率に応ずる)し、完全培地を用いてさらに 48 時間培養する。
7. 死滅曲線より決定した適切な抗生物質の最低濃度を利用して、感染細胞より定常発現細胞を選択できる。その後、ウェスタンブロット、配 列決定、surveyor アッセイといった種々の技術を利用し、ゲノム編集アッセイを行うことができる。
注: VSV-G 糖タンパク質発現は 293T 細胞の融合の原因となるため、融合細胞として知られる大型の複数核細胞様式を示す。この形態 変化は正常であり、レンチウイルス産生には影響しない。
■ 増幅プロトコール
1. 組換え型アデノウイルスを入手後、2 ~ 3 に分注して1つを 293 細胞内での増幅に使用する。他の分注品はシードストックとして -70℃で保存する。
2. アデノウイルスを 60 ~ 70% の細胞密度の HEK 293 細胞で増幅する。60mm ディッシュでは 70 μ L のアデノウイルス、
100mm ディッシュでは 200 μ L のアデノウイルスを使用して細胞感染を行う。
3. 95% 以上の 293 細胞がディッシュより剥離した際、細胞と培地の両方を大きめのコニカルチューブに回収する。
4. 回収した溶液を -70℃の冷凍庫かドライアイス/エタノール内で凍結後、37℃のウォーターバスで溶解する。この凍結溶解を 3 回繰り返す。
5. 3,000rpm で 10 分間、室温で遠心し、細胞片を沈渣させる。
6. 上清を新しいチューブに移す。すぐに使用する場合(2 ~ 3 週間程度)は 4℃で保存し、長期保存する場合はグリセロールを最終濃度 10% となるように加え -70℃で凍結する(1 ~ 2 年程度安定)。
7. トランスダクション手順:
ウイルスをin vitro トランスダクションに使用する場合、ほとんどのヒト細胞株においてウイルス上清がほぼ 100% の導入効率を示 すため、ウイルス上清を 2 回繰り返して CsCl 精製する必要はない。in vivo 実験の場合には、欠損粒子、細胞片および残余する培地成 分により顕著な免疫応答誘導が生ずることがあるため、これらを除去するために CsCl 精製は必須となる。さらに、CsCl 精製によりin vivo 注入に適したレベルのウイルス濃度に濃縮することができる。
1. トランスダクション前日に、標的細胞を 70% の細胞密度となるよう 6 穴プレートまたは 10cm ディッシュに播種する。
2. 培養培地を吸引除去し、ウイルス培養上清(6穴プレートには 1mL, 10cm ディッシュには 4-5mL)を加えて細胞を覆い、培養器 内で1時間培養する。
3. ウイルス含有培地を除去し新しい完全培地と交換する。
4. ゲノム編集はトランスダクションから 48 ~ 72 時間後にウェスタンブロット、配列決定または Surveyor アッセイなどにより評 価することができる。
重要情報
アデノウイルスシードストックは可能であれば1つずつ個別に異なる滅菌ベンチや培養装置を用いて増幅することを推奨します。もし、こ れら機器が1つしかない場合、各ウイルスを経時的に増殖するとともに、各ウイルスを使用した後に30分間のUV照射を行います。2種類以 上のアデノウイルスを同時に使用することが相互汚染の主な原因となることから、ウイルスごとに異なるトリプシンや培地容器のご利用 をお奨めします。
Applied Biological Materials 社の構築済みアデノウイルスはシードストックとしてのみご提供しています。そのため、in vitro トランスダ クション等を行う前に増幅して頂く必要があります。in vivo 用には大容量でのウイルス産生と精製が必要となります。
構築済みアデノウイルスの増幅
DNA2.0 社
Santa Cruz Biotechnology 社 技術情報
GeneCopoeia 社 OriGene Technologies 社 技術情報 DNA 2.0 社 技術情報 Applied Biological Materials 社 技術情報 CRISPR- Cas システム 概論 ゲノム編集総説
DNA2.0 社 CRISPR/Cas9 ゲノム編集 All-in-one ベクター
独自の NickaseNinja All-in-One ベクターで CRISPR/Cas9 技術を用いたゲノム 編集をさらに簡単に!
ゲノム編集(Genome Editing)とは、CRISPR/Cas システムや Transcription Activator- Like Effector Nucleases(TALEN)等の技術により遺伝子特異的な破壊やレポーター遺伝 子のノックイン等を行う新しい遺伝子改変技術です。DNA2.0 社では、CRISPR/Cas9 ベク ター、試薬、gRNA デザインソフトを含む、ゲノム編集操作用ツールセットを開発しました。画期 的な NickaseNinja™ は、単一ベクターから2つの gRNA をタンデム発現し、本品1つで全操 作が完了する All-in-One ベクターです。CRISPR/Cas9 技術の利用により複雑なゲノムを的確 に改変することが可能です。DNA 社は、無料でご利用頂ける gRNA デザインツールを WEB 上(https://www.dna20.com/eCommerce/cas9/input)にご用意しており、これを用いて gRNA をデザインし、Electra™-CRISPR ベクターにクローニングします。
特長
CRISPR/Cas9 技術
● 特定領域への遺伝子挿入
● 特定部位における遺伝子発現のノックアウト
● 簡便: Electra™ システムで gRNA のカセットクローニング E.coli、酵母、
および哺乳類細胞ゲノムに対応
NickaseNinja™ オールインワンベクター
● 高精度、オフターゲット作用を低減
● 簡便、単一ベクターの形質転換
● Nikase ベクターへのタンデム gRNA クローニング
● 2A- 結合型レポータより選択: DasherGFP, PaprikaRFP、レポータなし
● 最小限のオフターゲット作用 DNA
WEBへどうぞ
記事ID:12575
DNA2.0 社
CRISPR/Cas9 ゲノム編集 All-in-one ベクター
使用目的
● 特定領域への遺伝子挿入
● 特定部位における遺伝子発現のノックアウト
● E.coli 、酵母、および哺乳類細胞のゲノム編集操作 CRISPR/Cas9 ベクターの特長
● NickaseNinja™ の利用
単一ベクターに 2 つの gRNA がタンデムで組込まれ、トランスフェクション効率が増大
● プロモーターを選択可能:CMV / CBh / CAG
● 二本鎖または一本鎖切断
Cas9 (S. pyogenes) は二本鎖切断を行います。一方、Cas9N ニッカーゼ変異体は一本鎖切断を行います(特異性をより向上させるため には、2 つのタンデム gRNA をもつ Nickase Ninja™ ベクターを利用*1(15 ページ参照))。
Cas9 ヌクレアーゼは 20 塩基のガイド配列により誘導され、ほとんど全てのゲノム座位を切断可能です。これにより生じた染色体の破損 は、通常、非相同末端結合(NHEJ)により修復されるため、標的座位にわずかな欠損や挿入が生じます。一方、相同ドナー DNA を Cas9 とトランスフェクションした場合、相同組換え修復システムが刺激され標的配列を期待する変異配列と置換できます。
Cas9 には、DNA の非相補鎖を切断する RuvC 様ヌクレアーゼドメインと相補鎖 HNH ヌクレアーゼドメインという 2 つの触媒領域が あります。DNA2.0 社の Cas9N またはニッカーゼは、Cas9 ヌクレアーゼの RuvC 様ヌクレアーゼドメインの D10A 点変異体です。
ガイド RNA は、ワトソン・クリック塩基対により Cas9 をゲノム標的に導くため、いかなるゲノム座位をも簡単に標的できます。
プロモータに関して特にご希望がない場合、または DNA2.0 社で検証済みの細胞(HeLa, HEK293, CHO および神経細胞)以外の細胞 をご利用の場合、3 種を全てお試し頂くことをお奨めします。
CRISPR/Cas9 参考文献
● オフターゲット作用低減かつ挿入欠損効果増大に向けた Cas9 ニッカーゼ戦略に関する報告
Science 2013. 339(6121):819. Multiplex Genome Engineering Using CRISPR/Cas Systems.
● ゲノム編集用 CRISPR/Cas9 システムに関する報告
Science 2013. 339(6121):823. RNA-Guided Human Genome Engineering via Cas9.
図 1. CRISPR/Cas9 システム作用機序概要