ゲノム編集ツール
Edit-R
™
CRISPR-Cas9
テ
ク
ニ
カ
ル
・
マ
ニュ
ア
ル
imagination at work
Dharmacon
™
(ダーマコン)
RNAi
、遺伝子発現、
ゲノム編集
細菌・古細菌の適応免疫防御機構、
CRISPR-Cas ...3
CRISPR-Cas9
システムの改変、哺乳動物のゲノム編集への応用
...3
2 Edit-R CRISPR-Cas9
ゲノム編集ツール
... 4
Edit-R SMARTCas9 Expression Plasmid
(
Cas9
ヌクレアーゼ発現用プラスミド)
...4
Edit-R trans-activating CRISPR RNA
(
tracrRNA
)
...5
Edit-R CRISPR RNA
(
crRNA
)
...5
crRNA
の設計について
...6
設計ツール
Dharmacon CRISPR RNA Configurator
による
crRNA
の設計
...6
カスタム
crRNA
のご注文
...6
3 Edit-R CRISPR-Cas9
ゲノム編集ツール各コンポーネントによるコトランスフェクション
... 7
必要な
Edit-R CRISPR-Cas9
ゲノム編集ツール関連材料
...7
その他の必要な材料
...7
Edit-R CRISPR-Cas9
ゲノム編集ツール各コンポーネントによるコトランスフェクションの
一般的な手順
...8
細胞の播種
...8
コトランスフェクション
...8
ゲノム編集によって変異を導入した細胞の濃縮
...9
ゲノム編集結果の確認アッセイに関する推奨事項
...10
4
トランスフェクション条件の最適化
...10
5 Appendix ...12
ブラストサイジン耐性マーカー
Blast
R搭載
Edit-R CRISPR-Cas9 Nuclease Expression Plasmid ...12
安定性と保存
... 13
Dharmacon Edit-R SMARTCas9 Expression Plasmid ... 13
Dharmacon Edit-R tracrRNA
、
crRNA ... 13
Dharmacon DharmaFECT Duo Transfection Reagent ... 13
FAQ
(よくある質問)
...14
文献
...18
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル
1
CRISPR-Cas9
システムによるゲノム編集の概要
細菌・古細菌の適応免疫防御機構、
CRISPR-Cas
CRISPR(clustered regularly interspaced palindromic repeats)-Cas(CRISPR-associated proteins)システムは細菌や古細菌 が備える適応免疫性の防御機構であり、外来の核酸を認識しその機能を阻害する働きがあります。細菌や古細菌は、バクテリオ
ファージなど外来のDNA因子に感染を受けて宿主となるとき、その外来DNA因子から(プロト)スペーサーと呼ばれる短い配列
を切り取り、自身がもつゲノムの特定領域(CRISPR遺伝子座)にパリドローム(回文配列)様リピート配列とともに組み込みます。
さまざまなスペーサーとリピート配列からなる複数のユニットがCRISPR遺伝子座に集まり、クラスター化することにより、CRISPR
配列と呼ばれる配列が形成されます。CRISPR配列を含む遺伝子座全体がRNAポリメラーゼにより転写されてプレCRISPR RNA(プ レcrRNA)と呼ばれる一次転写産物を生じ、このプレcrRNAがさらにCRISPR RNA(crRNA)と呼ばれる短い成熟型のRNAに分 割されます。このとき得られるcrRNAには、外来DNA因子に対して相補的な配列が含まれています。crRNAは多機能性のタンパ ク質またはタンパク質複合体(CRISPR-associated[Cas]タンパク質)を呼び込み、外来DNA中の短鎖protospacer-adjacent motif(PAM)に隣接する相補的な標的配列を切断します。これにより、宿主菌は感染の進行から守られます(Bhaya, 2011)。
CRISPR-Cas9
システムの改変、哺乳動物のゲノム編集への応用
細菌や古細菌のCRISPR-Casシステムとしてはこれまでに多くの種類が同定されており、そのメカニズムやCasタンパク質、サブ ユニットが形成する複合体にも様々な種類があることが確認されています。特に
Streptococcus pyogenes
(S. Pyogenes
)がも つCRISPR-Cas9システムに関してはそのプロセスや主なコンポーネントの研究が進んでおり、このシステムを改変して哺乳動物細 胞のゲノム編集に応用する試みがなされています。S. pyogenes
がDNAのPAMに隣接する特定配列を標的とし、これを切断する プロセスは、わずか3つのコンポーネントにより行われます(Jinek, 2012)。(1)Cas9エンドヌクレアーゼ
(2)crRNA:CRISPR遺伝子座(CRISPR配列)の転写により生じる成熟型RNA
(3)trans-activating CRISPR RNA:CRISPR遺伝子座から生じるもうひとつのRNAであり、crRNAと部分的にハイブリッドを形成 する
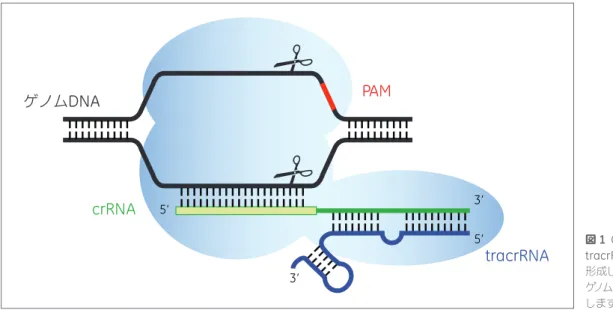
図1に模式図を示しています(tracrRNA、Deltcheva, 2011)。
哺乳動物の細胞は、部位特異的な二本鎖DNAの切断を受けた場合にそれを修復する機能を備えています。修復は非相同末端結
合(NHEJ)か相同組み換え(HR)かのいずれかの過程により行われますが、NHEJによる修復は短鎖の挿入や欠失を伴う不完全 なものであることが多く、これらの挿入や欠失がナンセンス変異を生じてその遺伝子の破壊、ノックアウトに至る場合があります (Mali, 2014; Sampson, 2014)。細胞自体が行うこのDNA切断修復過程と、
S. pyogenes
のCRISPR-Cas9システムという非常に扱いやすい系とを組み合わせることで、シンプルなプラットフォームによる遺伝子機能の恒常的な破壊が可能になります。 ゲノム
DNA
PAM
tracrRNA
crRNA
5’ 3’ 5’ 3’ 図1 Cas9ヌクレアーゼの模式図(水色)。 tracrRNA(青)とcrRNA(緑)との複合体を 形成し、crRNAのもつガイド配列と相補的な ゲノムDNAの両鎖をPAM(赤)の5'側で切断 します。2 Edit-R CRISPR-Cas9
ゲノム編集ツール
Edit-R CRISPR-Cas9 ゲノム編集ツールは哺乳動物細胞のゲノム編集に必要な3つのコンポーネントから構成されています。 (1)Cas9ヌクレアーゼを発現する遺伝子配列を含むプラスミド(哺乳動物での発現用にコドン最適化済み) (2)74塩基長のtracrRNA (3)ご希望の標的配列に合わせて設計したcrRNA いず れも天 然 のS.pyogenesが もつCRISPR-Cas9システムをベースとしたコンポーネントで、 トランスフェクション 試 薬DharmaFECT ™ Duo Transfection Reagentを使ってこれら3つのコンポーネントすべてを同時に、目的の哺乳動物細胞に導入
することにより、遺伝子の破壊を行います。実験の全体的なワークフローは図2のとおりです。以下、個々のコンポーネントにつ いて詳しく説明します。 ゲノムが編集された細胞を
FACS
法で濃縮 ゲノムが編集された細胞を 抗生物質選択で濃縮 SMARTCas9 Expression Plasmid (mKate2)使用時 SMARTCas9 Expression Plasmid (PuroR)使用時 納品後 培養2
~3
日間 ターゲットcrRNA
の設計 crRNAの配列を 1遺伝子あたり3~5種類、 カスタム設計します Cas9ヌクレアーゼ 発現用プラスミド tracrRNA:crRNA 1. カスタム設計したcrRNA 2. tracrRNA 3. Cas9ヌクレアーゼを発現 用プラスミド 4. DharmaFECT Duo トランスフェクション試薬Web
で注文 トランスフェクション 培養3
~4
日間 図2 Edit-R CRISPR-Cas9 ゲノム編集ツールを用いた 遺伝子ノックアウトのワークフローEdit-R SMARTCas9 Expression Plasmid
(
Cas9
ヌクレアーゼ発現用プラスミド)
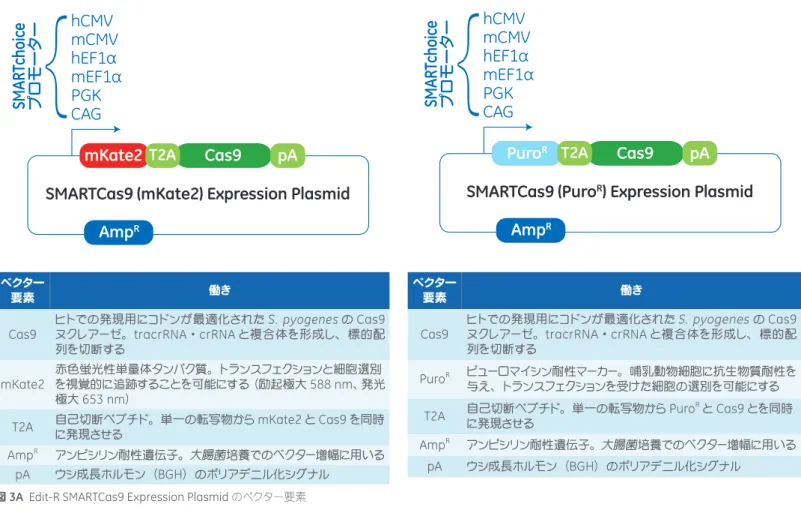
S. pyogenesのCas9(Csn1)遺伝子をヒトでの発現用に最適化した配列、蛍光レポーター(mKate2、Evrogen社、モスクワ)かピュー ロマイシン耐性マーカー(PuroR)かのいずれか一方、そしてこれらの上流に位置する単一のプロモーターを含む、Cas9ヌクレアー
ゼ発現用プラスミドです(図3A)。Cas9コード領域の3'末端ではなく5'末端にmKate2またはPuroRを配置することにより、C
末端に余分なアミノ酸が結合することによるCas9の活性低下を防いでいます。
プロモーターは複数のオプションのなかから、使用する細胞に合わせてもっとも活性が高いものをお選びいただくことが可能です (図3B)。SMARTCas9 Expression Plasmidは全製品とも、エンドトキシンを含まない乾燥状態のプラスミドDNAとなっており、
そのままトランスフェクションにお使いいただけます。
dharmacon.gelifesciences.co.jp
>
ゲノム編集
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル
このほか、プロモーターhCMVとブラストサイジン耐性マーカーを備えたCas9発現用プラスミドもご用意しています(ベクターマッ
プ、プラスミドの使用に際しての推奨事項に関しては12ページ「Appendix」をご覧ください)。
Edit-R trans-activating CRISPR RNA
(
tracrRNA
)
公表されている
S. pyogenes
のtracrRNA配列(Jinek, 2012)をベースに化学的に合成し、HPLC法で精製した長鎖RNAです。 複数の哺乳動物細胞で効率的なゲノム編集を行えることが試験で確認されています。Edit-R CRISPR RNA
(
crRNA
)
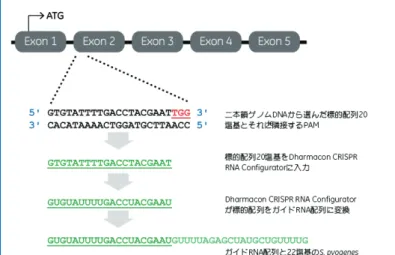
切断するゲノムDNAの標的配列(プロトスペーサー)と同じ配列の20塩基、そしてtracrRNAと相互作用をもつ特定の
S.
pyogenes
由来リピート配列からなる合成RNAです。プロトスペーサーとするゲノムDNA中の標的配列としては、そのゲノムDNAがもつPAMの上流側近傍の配列をお選びいただく必要があります。
S. pyogenes
の主要なPAM塩基配列はNGGです(図4)。図3B Edit-R SMARTCas9 Expression Plasmidに 使用できるプロモーター一覧
SMARTCas9 (mKate2) Expression Plasmid
hCMV
mCMV
hEF1α
mEF1α
PGK
CAG
mKate2
Amp
RSMAR
Tchoice
プ
ロ
モ
ー
ター
Cas9
SMARTCas9 (Puro
R) Expression Plasmid
hCMV
mCMV
hEF1α
mEF1α
PGK
CAG
Puro
RAmp
RSMAR
Tchoice
プ
ロ
モ
ー
ター
pA
T2A
Cas9
pA
T2A
SMARTCas9 (mKate2) Expression Plasmid
hCMV
mCMV
hEF1α
mEF1α
PGK
CAG
mKate2
Amp
RSMAR
Tchoice
プ
ロ
モ
ー
ター
Cas9
SMARTCas9 (Puro
R) Expression Plasmid
hCMV
mCMV
hEF1α
mEF1α
PGK
CAG
Puro
RAmp
RSMAR
Tchoice
プ
ロ
モ
ー
ター
pA
T2A
Cas9
pA
T2A
ベクター 要素 働き Cas9 ヒトでの発現用にコドンが最適化されたS. pyogenesのCas9 ヌクレアーゼ。tracrRNA・crRNAと複合体を形成し、標的配 列を切断する mKate2 赤色蛍光性単量体タンパク質。トランスフェクションと細胞選別 を視覚的に追跡することを可能にする(励起極大588 nm、発光 極大653 nm)T2A 自己切断ペプチド。単一の転写物からに発現させる mKate2とCas9を同時 AmpR アンピシリン耐性遺伝子。大腸菌培養でのベクター増幅に用いる
pA ウシ成長ホルモン(BGH)のポリアデニル化シグナル
図3A Edit-R SMARTCas9 Expression Plasmidのベクター要素
ベクター 要素 働き Cas9 ヒトでの発現用にコドンが最適化されたS. pyogenesのCas9 ヌクレアーゼ。tracrRNA・crRNAと複合体を形成し、標的配 列を切断する PuroR ピューロマイシン耐性マーカー。哺乳動物細胞に抗生物質耐性を 与え、トランスフェクションを受けた細胞の選別を可能にする
T2A 自己切断ペプチド。単一の転写物からに発現させる PuroRとCas9とを同時 AmpR アンピシリン耐性遺伝子。大腸菌培養でのベクター増幅に用いる pA ウシ成長ホルモン(BGH)のポリアデニル化シグナル プロモーター 説明 hCMV ヒトサイトメガロウイルス前初期プロモーター mCMV マウスサイトメガロウイルス前初期プロモーター hEF1α ヒト伸長因子1αプロモーター mEF1α マウス伸長因子1αプロモーター PGK マウスホスホグリセリン酸キナーゼプロモーター CAG トリβアクチンハイブリッドプロモーター
crRNA
の設計について
crRNAは目的の標的配列に合わせて設計することが可能です。次の手順に従ってください。
• 切断したいゲノムDNA配列のなかから、PAM(
S. pyogenes
の場合はNGG)の5'側(上流側)近傍に位置する塩基20個を 選択します。• 先頭の塩基の種類は、切断したいゲノムDNA配列に含まれているものであればどれでもかまいません。RNAの転写開始にU6
プロモーターを用いる系とは異なり、5'末端の塩基がGである必要はありません。
• 標的配列は切断したいゲノムDNA配列のどちらの鎖にも設定することが可能です(図4)。
• 選択した標的配列と同一の配列や酷似する配列がPAMの5'側近傍、特に同じゲノムDNA配列内の他のコード領域に存在する ことがないよう、配列の照合を行います(Fu, 2013; Hsu, 2013; Wang, 2013)。
PAMから3塩基分上流側の位置で、DNAの両鎖がCas9ヌクレアーゼにより切断されます。
設計ツール
Dharmacon CRISPR RNA Configurator
による
crRNA
の設計
• crRNAを 設 計するため のツールとしてDharmacon CRISPR RNA Configuratorをご 利 用 いた だ けます(dharmacon.
gelifesciences.com/gene-editing/edit-r/custom-crrna/をクリックし、"I want to design a crRNA"[crRNAを設計する]
と書かれたタブを開いて遺伝子のデータを入力してください)。ツールが次の手順により遺伝子ノックアウト実験用のcrRNA を設計します。 • 目的の遺伝子のなかから、その遺伝子の機能を喪失させる可能性のある標的配列を特定します。標的配列の検索は遺伝情 報を含む転写物の全バリアントを対象に、その遺伝子を構成する最上流のエクソンから順次行われます。検索対象を特定の バリアントのみに絞り込むことも可能です。 • 特異性向上のため、配列の照合を行います。選択した標的配列がPAMの5'側(上流側)近傍の配列と照合され、同一の配 列や酷似する配列があればそれが検出されます。同じ遺伝子内の他の領域と完全に一致している、あるいは一致率が非常に 高いcrRNA配列は排除されます。
カスタム
crRNA
のご注文
• 標 的 配 列 に設 定する20塩 基をすでにお決 めに なっている場 合 は、 カスタムcrRNAの 合成 をご注 文いただ けます。dharmacon.gelifesciences.com/gene-editing/edit-r/custom-crrna/にアクセスし、"Use my own crRNA sequence"(自
分のcrRNA配列を使う)と書かれたタブを開いて配列を入力してください。
• 個々の標的配列について20塩基の情報を入力してください。Dharmacon CRISPR RNA Configuratorによって所定の
S.
pyogenes
由来リピート配列が3'末端に付加され、42塩基のカスタムcrRNAが完成します(図4)。図4 ヒト遺伝子PPIB(chr15:64448014∼64455354)のノックアウトを行 うための標的配列20塩基を選択する場合の例。PAM(下線、赤)の5'末端に 隣接する標的配列を選択してcrRNAに組み込みます。選択した20塩基の配列 (下線、緑、太字)をDharmacon CRISPR RNA Configuratorに入力すると、
DNA配列が自動的にRNA配列へと変換されるとともに、22塩基の長さをも つ所定のS. pyogenes由来リピート配列(緑)が3'末端に付加され、目的の 遺伝子に特異的な42塩基のcrRNAが完成します。完成したcrRNAをカート に入れてご注文ください。弊社にて合成を行います。
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル
3 Edit-R CRISPR-Cas9
ゲノム編集ツール各コンポーネントによる
コトランスフェクション
Edit-R CRISPR-Cas9 ゲノム編集ツールを構成する3つのコンポーネント(Cas9 Expression Plasmid DNA、tracrRNA、crRNA)に
よるコトランスフェクション、そしてそれに続く遺伝子ノックアウトを成功させるためには、トランスフェクション試薬DharmaFECT
Duo Transfection Reagentを使って入念な最適化を行い、処理する細胞の種類に合ったトランスフェクション条件を得ることが必
要となります。トランスフェクション条件の最適化に関する一般的な推奨事項については10∼12ページをご覧ください。以下の
手順は、これらの推奨事項に従って最適なトランスフェクション条件がすでに得られていることを前提としたものです。
必要な
Edit-R CRISPR-Cas9
ゲノム編集ツール関連材料
Edit-R CRISPR-Cas9 ゲノム編集ツール関連材料は弊社Webサイトdharmacon.gelifesciences.comにてお買い求めいただけます。
• Edit-R SMARTCas9 Expression Plasmid(プロモーターとレポーター、またはプロモーターとマーカー搭載の組合せから選択)
120 µg乾燥品
• SMARTCas9 Expression Plasmid、蛍光レポーターmKate2搭載
• Edit-R hCMV_mKate2-Cas9 Expression Plasmid DNA (cat #U-004100-120) • Edit-R mCMV_mKate2-Cas9 Expression Plasmid DNA (cat #U-004200-120) • Edit-R hEf1
α
_mKate2-Cas9 Expression Plasmid DNA (cat #U-004300-120) • Edit-R mEf1α
_mKate2-Cas9 Expression Plasmid DNA (cat #U-004400-120) • Edit-R PGK_mKate2-Cas9 Expression Plasmid DNA (cat #U-004500-120) • Edit-R CAG_mKate2-Cas9 Expression Plasmid DNA (cat #U-004600-120) • SMARTCas9 Expression Plasmid、哺乳動物細胞の抗生物質選択用マーカーPuroR搭載• Edit-R hCMV_PuroR-Cas9 Expression Plasmid DNA (cat #U-005100-120)
• Edit-R mCMV_PuroR-Cas9 Expression Plasmid DNA (cat #U-005200-120)
• Edit-R hEf1
α
_PuroR-Cas9 Expression Plasmid DNA (cat #U-005300-120)• Edit-R mEf1
α
_PuroR-Cas9 Expression Plasmid DNA (cat #U-005400-120)• Edit-R PGK_PuroR-Cas9 Expression Plasmid DNA (cat #U-005500-120)
• Edit-R CAG_PuroR-Cas9 Expression Plasmid DNA (cat #U-005600-120)
• Edit-R CRISPR Cas9 Nuclease Expression Plasmid、哺乳動物細胞の抗生物質選択用マーカーBlastR搭載、120 µg乾
燥品(Dharmaconカタログ番号:U-001000-120) ベクターマップ、プラスミドの使用に際しての推奨事項に関しては 「Appendix」をご覧ください。
• (オプション)トランスフェクション最 適 化用プラスミドmKate2 Transfection Optimization Plasmid、120 g乾燥 品(Dharmaconカタログ番号:U-003000-120) ベクターマップ、プラスミドの使用に際しての推奨事項に関しては 「Appendix」をご覧ください。
• tracrRNA、5 nmol(120 µg)乾燥品(Dharmaconカタログ番号:U-002000-120)
• crRNA、20 nmol乾燥品(Dharmaconカタログ番号:CTM-XXXXXX-XXX、dharmacon.gelifesciences.com/gene-editing/
edit-r/custom-crrna/で目的の遺伝子に合わせて設計、ご注文いただいたもの)
• トランスフェクション 試 薬DharmaFECT Duo Transfection Reagent(Dharmaconカタログ 番 号:T-2010-01[0.2 ml]、
T-2010-02[0.75 ml]、T-2010-03[1.5 ml]、T-2010-04[1.5 ml × 5本])
その他の必要な材料
細胞培養で標準的に用いられる試薬および細胞の維持に適した装置が必要になります。以下の材料、器具類を別途ご用意ください。
• 抗生物質を含まない完全培地。処理する細胞の維持および継代培養に適した細胞培養培地(血清かサプリメント、または両方 を含む)で、抗生物質が添加されていないもの
• CellTiter-Blue Cell Viability Assay(Promega社カタログ番号:G8081)など、細胞生存率を評価するためのアッセイ
• 細胞集団でゲノム編集イベントを検出するためのアッセイ(複数可)
• (オプション)抗生物質ピューロマイシン(Fisher Scientific社カタログ番号:BP2956-100)
• (オプション)抗生物質ブラストサイジンS(Fisher Scientific社カタログ番号:BP2647-25)
• 10 mM Trisヌクレアーゼフリーバッファー(pH 7.4)
Edit-R CRISPR-Cas9
ゲノム編集ツール各コンポーネントによるコトランスフェクションの一般的な手順
哺乳動物の培養細胞にトランスフェクション試薬DharmaFECT Duo Transfection Reagentを使ってCas9 Expression Plasmid DNA、tracrRNAおよびcrRNAを導入する一般的な手順を以下に示します。処理する細胞の種類に合わせて条件の入念な最適化
を行い(10ページ「トランスフェクション条件の最適化」を参照)、試薬の正確な量と添加条件を決めたうえで実験を開始してく ださい。なお以下の手順は、24ウェルプレートでHEK293T細胞のトランスフェクションを行う場合の条件を、あくまで一例として 示したものです。 すべての工程は細胞培養用層流フード内にて無菌法で行ってください。
細胞の播種
播種に最適な細胞数は細胞の増殖特性によって異なります。したがって、実験を行って決める必要があります。 1. 細胞をトリプシン処理し、細胞数をカウントします。 2. 抗生物質を含まない完全培地を加え、適切な培養開始密度まで希釈します。例えば24ウェルプレートで1ウェルあたり 100,000個のHEK293T細胞を播種する場合、細胞100,000個に対して培地0.5 mlの割合で希釈を行います。 3. 24ウェルプレートの各ウェルに細胞懸濁液0.5 mlを添加します。 4. 37℃、5% CO2の条件で終夜インキュベートします。コトランスフェクション
表1 ゲノム編集を目的としたコトランスフェクション実験に用いるサンプルの例 サンプル名 目的Cas9 Expression Plasmid DNAのみ ネガティブコントロール:標的を決めるゼを発現させる RNAがない状態でCas9 ヌクレアー
Cas9 Expression Plasmid DNA、tracrRNA、 目 的 の遺 伝 子に特 異 的 な
crRNA ゲノム編集サンプル:目的の遺伝子の二本鎖を意図した位置で切断するよ うRNAによってプログラムされたCas9 ヌクレアーゼを発現させる 未処理 未処理コントロールサンプル:細胞生存率を確認する 注: 試薬を添加した後にはピペッティングを行い、チューブの内容物を穏やかに撹拌してください。 5. ゲノム編集実験用として3種類の異なるサンプルを使用します(表1)。
6. pH 7.4の10 mM Trisバッファー(ヌクレアーゼフリー)1.2 mlをCas9 Expression Plasmid DNA 120 µgに加え、100 ng/ µlのCas9プラスミド希釈溶液とします。波長260 nmでの紫外分光法によりDNA濃度を確認し、必要であれば量を調節 して濃度を100 ng/µlとしてください。
7. pH 7.4の10 mM Trisバッファー(ヌクレアーゼフリー)500 µlをtracrRNA 5 nmolに加え、10 µMのtracrRNA希釈溶液 とします。波長260 nmでの紫外分光法によりRNA濃度を確認し、必要であれば量を調節して濃度を10 µMとしてください。
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル
8. pH 7.4の10 mM Trisバッファー(ヌクレアーゼフリー)2 mlをcrRNA 20 nmolに加え、10 µMのcrRNA希釈溶液とします。 波長260 nmでの紫外分光法によりRNA濃度を確認し、必要であれば量を調節して濃度を10 µMとしてください。
9. 1.7 ml容のチューブを使い、表2(2∼5列目)に従ってトランスフェクション用サンプルを調製してください。tracrRNA・
crRNA複合体の最終濃度は50 nM、Cas9 Expression Plasmid DNAの濃度は1ウェルあたり1 µgとなります。
10. 1 mg/mlのDharmaFECT Duo Transfection Reagent原液120 µlを無血清培地2 mlで希釈し、穏やかに混合すること により60 µg/mlのDharmaFECT Duo希釈溶液とします。常温で、チューブを5分間放置してください。
11. 表2(6列目)に従って各サンプルのチューブにDharmaFECT Duo希釈溶液50 µlを加えます。これにより、DharmaFECT Duoの最終濃度は1ウェルあたり3 µgとなります。トランスフェクションを行わないコントロール用サンプルは DharmaFECT Duo希釈溶液を加えず、無血清培地のみとしてください。各チューブとも、総液量は100 µlとなります。ピペッ ティングにより穏やかに撹拌した後、常温で20分間放置してください。 12. 抗生物質を含まない完全培地400 µlを各サンプルに加えて総液量を500 µlとします(7、8列目)。得られた液がトランスフェ クション用の培地となります。 13. 細胞が入った24ウェルプレートの各ウェルから培地を抜き、代わりに適切な種類のトランスフェクション用培地500 µlを各 ウェルに加えます。 14. 37℃、5% CO2の条件で細胞を48∼72時間にわたりインキュベートした後、ゲノム編集結果の確認に進みます。
ゲノム編集によって変異を導入した細胞の濃縮
Cas9によるゲノム編集の頻度が高い細胞集団を得るには2種類の方法があります。どちらの方法を用いるかは、SMARTCas9 Expression Plasmidのうちどちらの種類を実験に使ったかによって異なります。a. mKate2-Cas9 Expression Plasmid DNAを用いた場合:FACS法で濃縮
mKate2-Cas9 Expression Plasmid DNAのトランスフェクションから48∼72時間が経過した細胞を使用します。
一般的なFACS法を行える十分な数の細胞を回収できるよう、トランスフェクションには組織培養用の大型ディッシュ をお使いください。また、細胞はmKate2の発現量が少ないもの、中程度のもの、そして多いものという3つの画 分に分けることをおすすめします。下流のアプリケーションや試験のため必要な画分の細胞の増殖を簡単に行えるよ う、それぞれの画分には十分な数の細胞を割り当ててください。 表2 24ウェルプレートでのゲノム編集実験に用いるトランスフェクション用サンプルの調製 サンプル名 無血清培地 tracrRNA 希釈溶液 (10 µM) crRNA 希釈溶液 (10 µM) Cas9 プラスミド 希釈溶液 (100 ng/µl) DharmaFECT Duo 希釈溶液 (60 µg/ml) 成長培地 1 ウェルあたり の総液量
Cas9 Expression Plasmid DNAのみ 40 0 0 10 50 400 500
Cas9 Expression Plasmid DNA、tracrRNA、
目的の遺伝子に特異的なcrRNA 35 2.5 2.5 10 50 400 500 未処理 100 0 0 0 0 400 500 24ウェルのHEK293T細胞各サンプルに用いる量(µl)。複数のウェルを1組として処理を行う場合はウェルの数に応じ、またピペッティングのミスも考慮して、 十分な量を調製してください。他の種類の細胞でトランスフェクションを行う場合は、条件の入念な最適化を行い(10ページ「トランスフェクションの最適化」 を参照)、試薬の正確な量を決めたうえで実験を開始してください。 注:ゲノム編集結果の確認には細胞の一部をお使いください(次ページ「ゲノム編集結果の確認アッセイに関する推奨事項」を参照)。 編集結果の確認後、クローン細胞の生成を行える十分な数の細胞が残っている必要があります。 注:ゲノムの編集が検出できない、アッセイで測定した編集頻度が低いといった場合は細胞集団の濃縮をご検討ください。濃縮の 手順は次の項に記載しています。
mKate2は励起極大588 nm、発光極大633 nmの遠赤外蛍光性単量体タンパク質です。推奨されるフィルターは
Omega Optical社QMAX-RedとXF102-2のセット、またはその同等品ですが、Texas Redやそれに類するフィルター セットでの検出も可能です。
b. PuroR-Cas9 Expression Plasmid DNAを用いた場合:ピューロマイシンで選択
ピューロマイシンでの選択による濃縮にはPuroR-Cas9 Expression Plasmid DNAを導入した細胞を使用します。適す
るピューロマイシン濃度は細胞の種類によって異なりますので、選択を始める前に死滅曲線を使って実験的に決定する 必要があります。トランスフェクションから48∼72時間が経過した細胞を、選択の開始前に分割してお使いください。 選択培地中で細胞をさらに2∼4日間、インキュベートします。 抗生物質は、活発に分裂している細胞に対して最大の効果を発揮します。そのため、コンフルエンスが25%を超えな いよう細胞を分割することが望ましいといえます。
ゲノム編集結果の確認アッセイに関する推奨事項
細胞集団での挿入・欠失の検出方法としてもっともよく使われているのは、SURVEYOR® Mutation Detection Kit(Integrated
DNA Technologies社)やT7 Endonuclease(Guschin, 2010; Reyon, 2012; Cong, 2013)といったミスマッチ検出アッセイです。
これらのアッセイはいずれも、精製ゲノムDNAと細胞溶解液の両方に対応しています。 ゲノムが編集された細胞の増殖によりクローン細胞集団を得る場合の編集結果の確認方法としてはサンガー法がもっともよく使わ れています(Reyon, 2012)。
4
トランスフェクション条件の最適化
処理する細胞の種類に合わせてトランスフェクション条件を入念に最適化することで、Edit-R CRISPR-Cas9 ゲノム編集ツール各 コンポーネントのトランスフェクション効率を最大限に高め、かつ細胞生存率への影響を最小限に抑えることが可能になります。 トランスフェクション条件の最適化では、処理する細胞に対してもっとも強い活性を示すプロモーターをEdit-R mKate2-Cas9 Expression Plasmid DNAに搭載したプラスミドを使って、蛍光強度(励起極大588 nm、発光極大633 nm)が最大となりかつ細胞生存率が未処理細胞との比較で70%を超える条件を特定します。96ウェルプレートで簡単な方法により行えますので、少量
のプラスミドで複数のトランスフェクション条件を同時にご検討いただけます。特定した条件は表面積に基づいてスケールアップし、 より規模の大きい組織培養用ディッシュでのトランスフェクションに適用することが可能です。
注:トランスフェクション試薬DharmaFECT Duo Transfection ReagentはRNAとプラスミドの両方を細胞に導入する用途に 最適化されています。他のトランスフェクション試薬をお使いになる場合は、その試薬でRNA、プラスミドの両方を目的の 細胞に効率的に導入できるかどうかをあらかじめご確認ください。
最適化実験には少なくとも2種類の細胞密度(コンフルエンス60∼80%)、そして互いに濃度の異なる複数のDharmaFECT Duo Transfection Reagentが必要になります。推奨される各コンポーネントの濃度範囲は以下のとおりです。
• DharmaFECT Duo Transfection Reagent、20∼80 µg/ml(96ウェルプレートの1ウェルあたり0.2∼0.8 µlに相当)
• プラスミド、96ウェルプレートの1ウェルあたり100∼200 ng
以下、96ウェルプレートで3ウェル1組の実験を行うことによりトランスフェクション条件を最適化する場合の手順を説明します(表
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル 1. トランスフェクションを行う日のコンフルエンスが60∼80%となるよう、前日に組織培養用の96ウェルプレートに細胞を播 種します。少なくとも2種類の細胞密度を検討するようにしてください。 2. pH 7.4の10 mM Trisバッファー(ヌクレアーゼフリー)1.2 mlをプラスミド120 µgに加え、100 ng/µlのCas9プラスミド希 釈溶液とします。波長260 nmでの紫外分光法によりDNA濃度を確認し、必要であれば量を調節して濃度を100 ng/µlとし てください。3. 7種類のDharmaFECT Duo希釈溶液(20∼80 µg/ml)をそれぞれ異なるチューブに調製します。1 mg/mlのDharmaFECT Duo Transfection Reagent原液10 µl、15 µl、20 µl、25µl、30 µl、35 µlおよび40 µlを無血清培地500 µlで希釈し、穏 やかに混合してください。DharmaFECT Duoの最終濃度は96ウェルプレートの1ウェルあたり0.2∼0.8 µgとなります。チュー ブを常温で5分間放置してください。 4. 深型のプレートの各ウェルに無血清培地とCas9プラスミド希釈溶液を総液量が10 µlとなるように添加します(表4)。次に DharmaFECT Duo希釈溶液10 µlを加え、各ウェルに総液量20 µlのトランスフェクション用混合培地を形成します。それぞ れのDharmaFECT Duo希釈溶液でこの操作を繰り返します。ピペッティングにより穏やかに撹拌した後、常温で20分間放 置してください。トランスフェクションを行わないコントロール用ウェルはプラスミドやDharmaFECT Duo希釈溶液を加えず、 無血清培地のみとしてください。 表4 96ウェルプレートでのトランスフェクションの最適化に用いるサンプルの調製 サンプル名 無血清培地 Cas9プラスミド 希釈溶液 (100 ng/lµl) DharmaFECT Duo 希釈溶液 (20∼80 µg/ ml) 成長培地 1ウェルあたりの 総液量
Edit-R mKate2-Cas9 Expression Plasmid DNA 100 ng 9 1 10 80 100
Edit-R mKate2-Cas9 Expression Plasmid DNA 200 ng 8 2 10 80 100
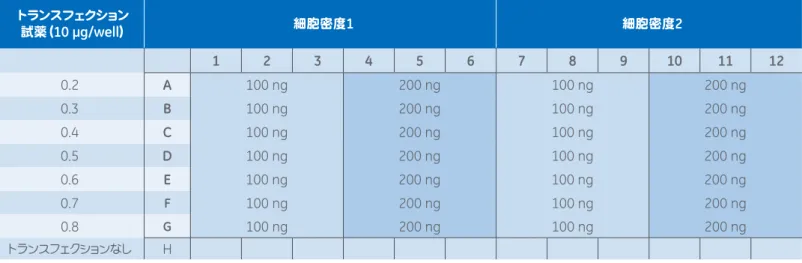
未処理 20 0 0 80 100 96ウェルの各サンプルに用いる量(µl)。96ウェルプレートの1ウェルあたりの量を示しています。3ウェルを1組として実験を行う場合は3つのウェルに十分な量を 添加できるよう、またピペッティングのミスも考慮して、すべての分量を3.5倍してください。 5. 工程4で得られた各サンプルに抗生物質を含まない完全培地80 µlを加えて総液量を100 µlとします。得られた液がトランス フェクション用の培地となります。 6. 細胞が入った96ウェルプレートの各ウェルから培地を抜き、代わりに適切な種類のトランスフェクション用培地(工程5で調 製したもの)100 µlを各ウェルに加えます。 トランスフェクション 試薬(10 µg/well) 細胞密度1 細胞密度2 1 2 3 4 5 6 7 8 9 10 11 12 0.2 A 100 ng 200 ng 100 ng 200 ng 0.3 B 100 ng 200 ng 100 ng 200 ng 0.4 C 100 ng 200 ng 100 ng 200 ng 0.5 D 100 ng 200 ng 100 ng 200 ng 0.6 E 100 ng 200 ng 100 ng 200 ng 0.7 F 100 ng 200 ng 100 ng 200 ng 0.8 G 100 ng 200 ng 100 ng 200 ng トランスフェクションなし H
太字のアルファベットはプレートの各行に、太字の数字は各列にそれぞれ対応しています。段階希釈したDharmaFECT Duo Transfection Reagentの各濃度を
A∼Gの各行で検討し、2種類の細胞密度をプレートの左半分と右半分で検討します。プラスミドの濃度は1ウェルあたり100 ngおよび200 ngの2種類とし、100 ngを1∼3列と7∼9列、200 ngを4∼6列と10∼12列で検討します。H行の細胞はトランスフェクションを行いません。
7. トランスフェクション実施後、mKate2発現量と細胞生存率を評価し、mKate2の発現量が最大でかつ細胞毒性の低い条件を 特定します。 a. トランスフェクションから24∼48時間後、細胞を顕微鏡で観察し、蛍光強度が最大となる条件を特定します。 b. トランスフェクションから48∼72時間後、細胞生存率アッセイを実施し、細胞毒性が最小限(細胞生存率が70%を超え ることが好ましい)に抑えられるDharmaFECT Duo濃度のうち最高の濃度を決定します。 注:細胞生存率は70%以上であることが理想ですが、これよりも細胞生存率が低いトランスフェクション条件で良好な編集効率 が得られる場合もあります。 注:最適化を行っても目的の細胞でのゲノム編集効率が低い場合は、 続くゲノム編集工程での濃縮をご検討ください。 濃縮の手順 は9ページ「ゲノム編集によって変異を導入した細胞の濃縮」に記載しています。
8. 特定した至適条件でSMARTCas9 Nuclease Expression PlasmidおよびtracrRNA・crRNA複合体のコトランスフェクション を行います。tracrRNA・crRNA複合体の濃度は25∼100 nMとしてください(通常は25 nMで編集効率が最大となります)。 最適化したトランスフェクション条件は組織培養の表面積に基づいてスケールアップすることが可能です。
注: mKate2が発する蛍光は、Edit-R mKate2-Cas9 Expression Plasmid DNAと同時に導入するtracrRNA・crRNA複合体の 濃度が高ければ高いほど弱くなります。
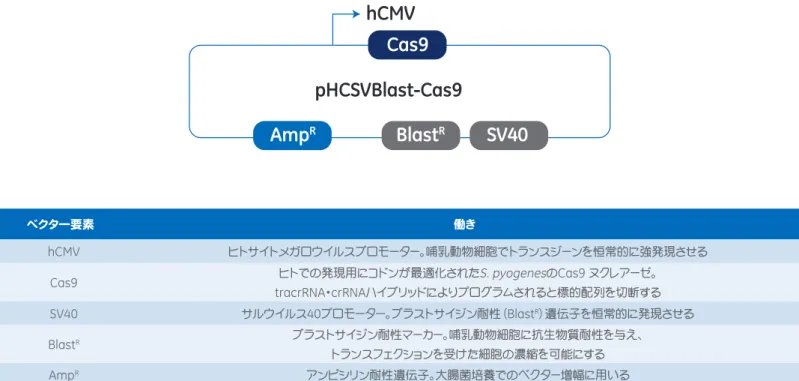
5 Appendix
ブラストサイジン耐性マーカー
Blast
R搭載
Edit-R CRISPR-Cas9 Nuclease Expression Plasmid
ヒトでの発現用にコドンが最適化された
S. pyogenes
のCas9(Csn1)遺伝子をヒトサイトメガロウイルス(hCMV)プロモーター から発現し、サルウイルス40(SV40)プロモーターの制御下でブラストサイジン耐性(BlastR)を発現する、ヌクレアーゼCas9 発現用プラスミドです(図5)。エンドトキシンを含まないプラスミドDNAとして、そのままトランスフェクションにお使いいただけ る量をお届けします。 ベクター要素 働き hCMV ヒトサイトメガロウイルスプロモーター。哺乳動物細胞でトランスジーンを恒常的に強発現させるCas9 ヒトでの発現用にコドンが最適化されたS. pyogenesのCas9 ヌクレアーゼ。
tracrRNA・crRNAハイブリッドによりプログラムされると標的配列を切断する
SV40 サルウイルス40プロモーター。ブラストサイジン耐性(BlastR)遺伝子を恒常的に発現させる
BlastR ブラストサイジン耐性マーカー。哺乳動物細胞に抗生物質耐性を与え、
トランスフェクションを受けた細胞の濃縮を可能にする
AmpR アンピシリン耐性遺伝子。大腸菌培養でのベクター増幅に用いる
図5 Edit-R BlastR-Cas9 Nuclease Expression Plasmidのベクター要素
pHCSVBlast-mKate
hCMV
mKate2
Blast
RSV40
Amp
RhCMV
Cas9
Blast
RAmp
RSV40
pHCSVBlast-Cas9
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル
ブラストサイジン耐性マーカーを搭載したEdit-R CRISPR-Cas9 Nuclease Expression Plasmidはブラストサイジン耐性を必要と
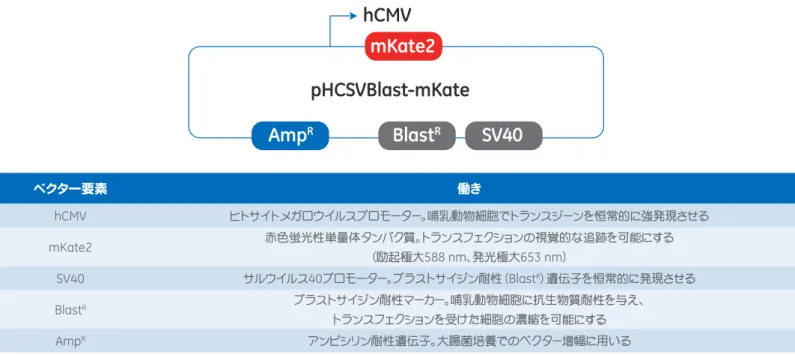
するゲノム編集実験にお使いいただけます。このほか互換性のあるプラスミドとして、ヌクレアーゼCas9を蛍光マーカーmKate2
で置き換えたトランスフェクション最適化用プラスミドmKate2 Transfection Optimization Plasmidもご用意しています(図6の ベクターマップを参照)。mKate2 Transfection Optimization Plasmidは10∼12ページ「トランスフェクション条件の最適化」 と同様の手順でトランスフェクション条件の最適化にお使いいただけます。
pHCSVBlast-mKate
hCMV
mKate2
Blast
RSV40
Amp
RhCMV
Cas9
Blast
RAmp
RSV40
pHCSVBlast-Cas9
Vector elements of the Edit-R Cas9 Nuclease Expression plasmid
ベクター要素 働き hCMV ヒトサイトメガロウイルスプロモーター。哺乳動物細胞でトランスジーンを恒常的に強発現させる mKate2 赤色蛍光性単量体タンパク質。トランスフェクションの視覚的な追跡を可能にする (励起極大588 nm、発光極大653 nm) SV40 サルウイルス40プロモーター。ブラストサイジン耐性(BlastR)遺伝子を恒常的に発現させる BlastR ブラストサイジン耐性マーカー。哺乳動物細胞に抗生物質耐性を与え、 トランスフェクションを受けた細胞の濃縮を可能にする AmpR アンピシリン耐性遺伝子。大腸菌培養でのベクター増幅に用いる
図6 Edit-R mKate2 Transfection Optimization Plasmidのベクター要素
安定性と保存
Dharmacon Edit-R SMARTCas9 Expression Plasmid
乾燥ペレットとして常温でお送りします。常温での使用期限は4週間です。お受け取り後すぐに-20∼-80℃で保管してください。
この保存条件で1年間は安定です。
保存の際には必ず、pH 7.4の10 mM Trisバッファー(ヌクレアーゼフリー)など、ヌクレアーゼを含まない溶液に溶解させた状 態としてください。
Dharmacon Edit-R tracrRNA
、
crRNA
乾燥ペレットとして常温でお送りします。常温での使用期限は4週間です。
お受け取り後すぐに-20∼-80℃で保管してください。この保存条件で1年間は安定です。
保存の際には必ず、pH 7.4の10 mM Trisバッファー(ヌクレアーゼフリー)など、ヌクレアーゼを含まない溶液に再懸濁させた 状態としてください。溶液を小分けにして-20℃で保存した場合2年間は安定です。
Dharmacon DharmaFECT Duo Transfection Reagent
保冷してお届けします。配送中に保冷剤が溶ける場合がございますが、製品への影響はございません。温暖な環境に長期間 置いても劣化しないことが安定性試験により確認されています(詳しくは弊社Webサイトdharmacon.gelifesciences.comの 「Service & Support」→「Documents & Downloads」→「Technical Resources」→「RNAi & カスタムRNA合成」より “Accell Stability Product Bulletin”をダウンロードしてご覧ください)。
FAQ
(よくある質問)
合成された
tracrRNA
および
crRNA
の品質管理はどのような試験で確かめられていますか。
Edit-R tracrRNAは、高速液体クロマトグラフィーを使って精製された後、MALDI-TOF質量分析とHPLCとを使って検証され、配
列長および純度が確認されています。また、crRNAも質量分析で検証されています。合成されたRNAの定量は、分光光度計を
用いて波長260 nmの吸光度を測定しています。
Dharmacon ™ 2
’
-ACE
法を用いて合成された
crRNA
は、ゲノム編集実験前に、高速液体クロマトグラフィーを使っ
て精製する必要がありますか。
Dharmacon ™ 2’-ACE法は、カップリング反応が極めて早く、高収量かつ高純度のRNAオリゴが得られます。脱塩処理を施さ
れ保護基が除去されたcrRNAをご提供しますので、ゲノム編集実験前に、新たに精製する必要はありません。弊社の検証試験では、 2位にある水酸基の脱保護処理を施されたcrRNAを使って良好なゲノム編集ができました。
バッファーを使って
crRNA
を再懸濁したところ、やや黄色味を帯びてしまいましたが、大丈夫でしょうか。
問題ありません。RNAオリゴの作製では、塩基の脱保護剤にはジチオレート系錯体を用います。少量の脱保護剤がサンプル中に 残存する場合があり黄色味を帯びていますが、ゲノム編集や細胞生存率に有害な作用を及ぼすことはありません。合成された
tracrRNA
および
crRNA
の安定性を教えてください。
乾燥したペレット状のRNAオリゴは、常温で、2∼4週間は安定ですが、長期の保存は、-20℃∼-80℃で冷凍します。この保存 条件では1年は安定です。無菌条件下で、リボヌクレアーゼやデオキシリボヌクレアーゼの無い環境で保存してください。Edit-R Plasmid
、
tracrRNA
および
crRNA
は凍結・解凍を何回繰り返して利用できますか。
4∼5回までは、凍結と解凍を繰り返しても、品質は保たれます。
tracrRNA
および
crRNA
は、どのように保存すれば良いでしょうか。
乾燥したRNAオリゴは、-20℃∼-80℃に設定された冷凍庫で保存しますが、霜がつかないようにします。pH 7.4のヌクレアーゼ フリーのバッファーを使って再懸濁すれば、凍結させたり解凍させたりしても安定です。RNAオリゴは再懸濁後に、小分けにして チューブに保存してください。4∼5回以上の凍結と解凍は繰り返してはなりません。分解が気になる場合は、ポリアクリルアミド ゲル電気泳動にて品質を確かめることができます。製品の配達が遅れたため、
Edit-R Cas9 Nuclease Expression Plasmid
、
mKate Transfection Optimization
Plasmid
、
tracrRNA
や
crRNA
が、一週間の間、常温下にありました。使用しても大丈夫でしょうか。
乾燥ペレットとして出荷されますので、常温下では、2∼4週間は安定です。RNAオリゴもプラスミドDNAも受け取ったら、直ち に、-20℃∼-80℃の冷凍庫で保管してください。
Edit-R Gene Engineering System
の各コンポーネントは、どのようにして出荷されるのですか。
Edit-R Cas9 Expression plasmid DNA、mKate2 Transfection Optimization plasmid DNA、tracrRNA、crRNAは、すべてが 乾燥ペレットとして、常温下で出荷されます。
RNA
は、分光光度計を使って定量しますが、その計算式を教えてください。
RNAの定量は、ベール法を用います:吸光度(260 nm)=ε×モル濃度×光路長(cm)。ここでεは、モル吸光係数を表します(crRNA のモル吸光係数は同梱された製品添付書に記載されております。また、tracrRNAのモル吸光係数は757400です)。計算式は、 モル濃度=(吸光度、照射度260 nm)/ε・ 光路長(cm)です。標準型の10 mmキュベットを使った場合、計算式において光 路長は1とします。それ以外のキュベットを使う場合、例えば2 mmのマイクロキュベットを使った場合は、光路長は0.2とします。Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアル
Edit-R Cas9 Expression Plasmid
の保存方法を教えてください。
乾燥プラスミドDNAとバッファーで再懸濁したDNAは、-20℃∼-80℃の場所で凍結保存します。凍結・解凍は繰り返さずに、 核酸の分解を防ぐために、リボヌクレアーゼやデオキシリボヌクレアーゼの無い環境で保存してください。
注文した
DharmaFECT Duo Transfection Reagent
が配達されてきたのですが、常温で放置してしまいました。試薬
は使っても大丈夫でしょうか。
DharmaFECT製品は、トランスフェクション試薬も含めて、多くの安定性試験および機能性試験を行った結果、たとえ保存条件 が悪くても、効率よくトランスフェクションができました。試験結果は、弊社Webサイトに掲載されておりますので、ダウンロー ドしてご利用ください(dharmacon.gelifesciences.com)。トランスフェクション後、細胞死が目立ちました。どのように対処すれば良いのでしょうか。
その場合は、導入条件を最適化する必要があります。考慮すべき条件は、トランスフェクション試薬に関しては、試薬の分量・試 薬のロット/バッチ、導入期間、また細胞に関しては、細胞培養継代数、細胞密度といった点があります。トランスフェクション 試薬濃度の低減や導入期間の短縮は、細胞への毒性作用を減らします。トランスフェクション試薬の作用は、ロット毎に異なるの で、そのために実験上のばらつきが起きることもあります。この問題が続く場合、トランスフェクション試薬を替えるか、テクニカ ルサポートにお問合せください(本書裏表紙を参照)。Edit-R Cas9 Gene Engineering
システムは、どのような細胞に用いることができますか。
本製品は、哺乳動物の細胞であれば導入が可能で、Cas9発現のためのプロモーターが作動するいかなる種類の細胞にもご利用
いただけます。
Edit-R Cas9 Expression Plasmid
にはいろいろな種類がありますがどれを使えば良いのでしょうか。
次の点を考慮してください。
1) ブラストサイジンを使ったCas9発現細胞の選択を希望し、使用する細胞内でヒトCMVプロモーターが作用する場合、哺
乳動物細胞の抗生物質選択用マーカーBlastR搭載のCas9ヌクレアーゼ発現プラスミドを使用できます。(コード番号:
U-0010000-120)
2) FACS装置を使って、蛍光マーカーを発現する細胞だけを選択する場合、SMARTCas9-mKate2 Expression Plasmidを使用
できます。このプラスミドは、6種の異なるプロモーターを選択できるので、使用する細胞でプロモーターの活性が強いもの
を選ぶことができます。
3) FACS装置を使わない場合、またはピューロマイシンを使う場合、SMARTCas9-PuroR Expression Plasmidを使用できます。
このプラスミドは6種の異なるプロモーターを選択できるので、使用する細胞内でプロモーターの活性が強いものを選ぶ ことができます。
Cas9
発現プラスミドと組合せる適切なプロモーターをどのように選べば良いのでしょうか。
弊社が提供するCas9プラスミドは、複数のプロモーターから選択することができます。これにより、Cas9ヌクレアーゼ遺伝子と mKate2レポーター遺伝子、あるいは、Cas9ヌクレアーゼ遺伝子とピューロマイシン耐性遺伝子を、効率よく発現できます。重 要なのは、実験観察に基づいてあるいは論文を参照したうえで、使用する細胞内で作用するプロモーターを搭載したEdit-R Cas9Expression Plasmidを使用することです。そのような情報が入手できない場合は、SMARTCas9-mKate2 Expression Plasmidを
Edit-R mKate2 Transfection Optimization Plasmid
の用途は?
トランスフェクション条件の最適化に使用します。ブラストサイジン耐性マーカーを組込んだEdit-R Cas9 Nuclease Expression Plasmidと構造が似ていますが、Cas9ヌクレアーゼ遺伝子がmKate2蛍光遺伝子配列に置き換わっています。Edit-R mKate2 Trasfection Optimization Plasmidを使ったトランスフェクション条件の最適化は、本書の「トランスフェクション条件の最適化」 の項に記述した手順に従ってください。ただし、SMARTCas9 Expression Plasmidを使用する場合、トランスフェクション条件の 最適化は、処理する細胞に対して活性を示すプロモーターを搭載したSMARTCas9-mKate2 Expression Plasmidを使います。
Edit-R tracrRNA
および
crRNA
は、他の
Cas9
ヌクレアーゼ発現プラスミドと組合せて使用できますか。
tracrRNAおよびcrRNAを使って検証した結果、哺乳動物細胞においてEdit-R Cas9 Expression Plasmidを使ったゲノム編集
は効率よく行えました。しかし、その他のCas9ヌクレアーゼ発現ベクターを使って実験した場合、編集効率は予測することが
できませんし、また問題が生じたときの解決方法を見出すこともできません。ただし、crRNAに含まれる繰り返し配列および
tracrRNAの全配列は、化膿連鎖球菌(Streptococcus pyogenes)のCRISPR-Cas9システムに由来するものなので、おそらく、他
のCas9ヌクレアーゼ発現プラスミドにも適用できると思いますが、そのためには、Cas9配列が正しく発現できるようコドンが
最適化されており、Cas9配列の発現は、使用する細胞で作用するプロモーターが制御する必要があります。しかも、プラスミド
DNAは、tracrRNAとcrRNAと共にコトランスフェクションを効率よく行う必要があります。
1
つのベクターから発現した単鎖ガイド
RNA
(
sgRNA
)よりも、2種類の合成された
RNA
(
tracrRNA
および
crRNA
)
を使う利点はどこにあるのでしょうか。
Edit-Rシステムはの利点は、複数のcrRNAを容易に入手し、使用できることです。Edit-Rシステムでは単一遺伝子の複数の標的
配列または複数遺伝子を容易にノックアウトできます。他方、sgRNAを一つのベクターより発現させる場合は、各標的配列を発
現ベクターにクローニングしなければならず、配列を確認しながらクローンを増殖・選別して、トランスフェクション用にDNAを分
離・精製します。この工程は、単一遺伝子の複数の標的または複数の遺伝子をノックアウトする場合、とても面倒です。
DharmaFECT Duo
とは異なるトランスフェクション試薬を使って、目的の細胞へ
Edit-R
のコンポーネントを導入して
もよいでしょうか。
DharmaFECT Duoは、プラスミドDNAおよびRNAオリゴを同時にトランスフェクションするために最 適化されています。
DharmaFECT Duoを用いてゲノム編集実験を行ったところ、Edit-Rコンポーネントの導入は効率良く行えることが分かりました。 その他のトランスフェクション試薬を使った場合のゲノム編集効率は予測することができませんし、また問題が生じたときの解決 方法を見出すこともできません。ただし、使用する細胞についてのコトランスフェクション条件が入念に最適化されている試薬で あれば、利用しても構いません。
DNA
ミスマッチ定量法では、ゲノム編集効率が低いのですが、なぜでしょうか。
Surveyor(IDT)やT7EI(NEB)など、ミスマッチを認識するDNAエンドヌクレアーゼを使った変異定量解析は、ゲノムDNAの対象と する配列をPCRで増幅した後、通常、ゲル電気泳動法を使って変異を検出します。CRISPR-Cas9を使って行ったゲノム編集にお ける塩基の挿入・欠失を正確に検出する方法は、使用するエンドヌクレアーゼによってその感度が異なり、以下の理由から、編集 効率が下がるように見えてしまいます。
1. 標的配列は、Cas9によって切断された後、非相同末端結合(NHEJ)によってDNAが修復された結果、さまざまなサイズの 欠失・挿入・変異が生じます。そのため、エンドヌクレアーゼによる切断後の電気泳動の結果では、ゲル泳動像が不鮮明でバ ンドがなかなか確認できなかったり、定量できなかったりします。 2. ミスマッチを認識するDNAエンドヌクレアーゼは、とりわけ反応時間を長くした場合、非特異的に作用してPCR産物を分解 することがあり、その場合、目的のバンドのシグナル強度が低下することがあります。 3. CRISPR-Cas9ゲノム編集によって大きな挿入や欠失が生じた場合、プライマー結合部位も影響を受けてしまい、変異を検出で きなくなることがあります。
Edit-R
と
SMARTCas9
を用いたゲノム編集
テクニカル・マニュアルピューロマイシン処理またはブラストサイジン処理を施すことで、ゲノム編集の効率は上がりますか。
ピューロマイシンやブラストサイジンは、編集された細胞だけを濃縮して、編集効率を上げますが、どのぐらい上がるかは導入条 件がどのぐらい最適化されているかによって異なります。Edit-Rコンポーネントのトランスフェクション効率が高いと、選択剤は編 集効率を若干向上させます。逆にトランスフェクション効率が低いと、選択剤は編集効率を大幅に向上させます。Edit-R
システムは、哺乳動物以外の例えば細菌や線虫といった下等生物の細胞での遺伝子ノックアウトに使用できま
すか。
Edit-R Cas9プラスミドは、哺乳動物の細胞で発現できるよう設計され、検証されています。哺乳動物以外の細胞を使った場合、 Edit-R RNAコンポーネントに関する編集効率は予測することができませんし、また問題が生じたときの解決方法を見出すことも できません。ただし、crRNAに含まれる繰り返し配列およびtracrRNAの全配列は、化膿連鎖球菌(Streptococcus pyogenes) のCRISPR-Cas9システムに由来するものなので、おそらく、他のCas9ヌクレアーゼ発現プラスミドにも適用できると思いますが、そのためには、Cas9配列が正しく発現できるようコドンが最適化されており、Cas9配列の発現は、使用する細胞で作用するプ
ロモーターが制御する必要があります。しかも、プラスミドDNAは、tracrRNAとcrRNAと共にコトランスフェクションを効率よ く行う必要があります。
Edit-R RNA
コンポーネントは、自分の使っている
Cas9
ヌクレアーゼ
mRNA
と組合せて使っても問題はありませんか。
また、変異
Cas9
ヌクレアーゼ発現プラスミドも使えますか。
tracrRNAおよびcrRNAを使って検証実験を行った結果、哺乳動物細胞においてEdit-R Cas9 Expression Plasmidを使ったゲ ノム編集は効率よく行えましたが、変異Cas9ヌクレアーゼ発現ベクターやCas9 ヌクレアーゼmRNAを使って実験した場合、編
集効率は予測することができませんし、また問題が生じたときの解決方法を見出すこともできません。ただし、crRNAに含まれ
る繰り返し配列およびtracrRNAの全配列は、化膿連鎖球菌(
Streptococcus pyogenes
)のCRISPR-Cas9システムに由来するものなので、おそらく、他のCas9コンポーネントにも適用できると思いますが、そのためには、Cas9配列が発現できるように
コドンが正しく最適化されており、しかも、酵素活性のあるCas9タンパク質が強く発現する必要があります。そのうえ、Cas9
mRNAまたはプラスミドDNAは、tracrRNAとcrRNAと共にコトランスフェクションを効率よく行う必要があります。
使用する細胞の倍数性は、
CRISPR-Cas9
によるゲノム編集結果に影響を与えますか。
CRISPR-Cas9システムを遺伝子の機能破壊に使う場合、対象とする遺伝子および細胞について十分な知識を持っていなければな りません。特に注意すべきことは、使用する細胞の倍数性、遺伝子のコピー数がいくつあるか、および一塩基変異多型(SNP)が 存在するかどうかです。野生型の二倍体細胞では、完全に遺伝子をノックアウトし表現型を観察するには対立遺伝子座の両方の 変異が必要です。がん細胞株と多くの不死化B細胞株は、染色体の異数性が見られるので、複数の遺伝子座が同時に変異する必 要があります。crRNAを設計する際に考慮すべきことは、SNPの有無とともに遺伝子座が複数あるかどうかということであり、そ れによって完全なノックアウトが実現できるかどうかが決まります。単一細胞に由来するクローン細胞を増殖させ、全ての遺伝子座 で必要な変異が得られているかの配列確認をすることが、完全なノックアウトを得るための間違いのない確認方法となります。文献
D. Bhaya, M. Davison,
et al
. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation.Annu
.Rev
.Genet
. 45, 273-297 (2011).L. Cong, F. A. Ran,
et al
. Multiplex Genome Engineering Using CRISPR/Cas Systems.Science
. 339(6121), 819-823 (2013). E. Deltcheva, K. Chylinski,et al
. CRISPR RNA maturation by trans-encoded small RNA and host factor Nuclease III.Nature
. 471(7340), 602-607 (2011).Y. Fu, J. D. Sander,
et al
. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs.Nat
.Biotechnol
. (2014). D.Y. Guschin, A. J. Waite,et al
. A rapid and general assay for monitoring endogenous gene modification.Methods Mol
.Biol
. 649, 247-256 (2010).F. Heigwer, G. Kerr,
et al
. E-CRISP: fast CRISPR target site identification.Nat
.Methods
. 11(2), 122-123 (2014).P.D. Hsu, D. A. Scott,
et al
. DNA targeting specificity of RNA-guided Cas9 nucleases.Nat
.Biotechnol
. 31(9), 827-832 (2013). M. Jinek, K. Chylinski,et al
. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity.Science
. 337(6096), 816-821 (2012).P. Mali, L. Yang,
et al
. RNA-guided human genome engineering via Cas9.Science
. 339(6121), 823-826 (2013).N. K. Pyzocha, F. A. Ran,
et al
. RNA-Guided Genome Editing of Mammalian Cells.Methods Mol
. Biol. 1114, 269-277 (2014) D. Reyon, C. Khayter,et al
. Engineering designer transcription activator-like effector nucleases (TALENs) by REAL or REAL-Fast assembly. Curr. Protoc.Mol
.Biol
. 100, 12.15.1‐12.15.14 (2012).T. R. Sampson, D. S. Weiss. Exploiting CRISPR/Cas systems for biotechnology.
Bioessays
. 36(1), 34-38 (2014).6 Evrogen
社ライセンスについて
Evrogen社の蛍光タンパク 本製品に含まれる、特許で保護された核酸配列は、蛍光タンパクをコードするものであり、特許の対象は、この蛍光タンパクの派 生物または改変物を含みます。米国および海外で特許出願中のもの、またEvrogen JSCが有する特許も対象となります(これら をまとめて、「Evrogen蛍光タンパク」と呼びます)。 本製品の購入により、買い手に譲渡不能ライセンスが移行し、①1買い手が学術団体または営利団体であるかに拘らず、非営利目 的での研究を行う場合(ここでいう研究とは、非商業的利用または活動を指し、その結果も含め、利益が生じないものとします。)、 および②2治療薬、診断用試薬、ワクチン、予防薬の開発に関わる検証を目的としたEvrogen蛍光タンパクによる評価を行う場合 に(ただし、これらの製品の製造または開発にEvrogen蛍光タンパクを使用しない場合に限ります。)、利用することができます。 いかなる目的であってもEvrogen蛍光タンパクの再販、配布、移譲、または第三者への利用権の提供、もしくは研究以外の目的 でかかる製品を利用することは、固く禁じられています。 買い手は、第三者に対し、かかる製品を使って作製した物質を販売、移譲、または商業目的で利用することはできません。買い手は、 商業目的としてではなく研究目的である場合に限り、本製品の利用によって得た情報を譲渡することができます。ここでいう商業 目的とは、①1製造における本製品の使用、②2本製品の使用によるサービス・情報・データーの提供、③3治療・診断・予防を目的 とした本製品の利用を含み、かつ対象となる団体のあらゆる活動を指します。 本製品の購入によって、Evrogen社のいかなる権利も化学分子の検証や検出を目的として移譲されることはなく、前述の特許ま たは特許出願におけるいかなる主張の下においてもライセンスが移譲されることはありません。 前述の特許、特許出願に関わる情報および上記で容認された以外の目的で本製品を利用するためにライセンスを購入する場合の 情報については、以下までお問合せください。お問合せ先:Licensing Department, Evrogen JSC
(住所:Miklukho-Maklaya street 16/10, Moscow,117997, Russian Federation、E-mai:[email protected])
Edit-R
と
SMARTCas9
を用いたゲノム編集
8
www.gelifesciences.co.jp
■機器アフターサービス
■納期/在庫お問合せ
注) お問合せに際してお客さまよりいただいた情報は、お客さまへの回答、弊社サービスの向上、弊社からのご連絡のために利用させて いただく場合があります。 注) アナログ回線等で番号選択ができない場合はそのままお待ちください。オペレーターにつながります。弊社 Web サイト ▲サポート ▲
FAQ
(製品 Q&A)または、
研究者向けコミュニティ
Life Sciences Academy
*▲▲