T H E W A K S M A N F O U N D A T IO N O F J A P A N IN C .
R e p o r t o f R e s e a r c h e s in 2 0 1 3 Dr. Selman A.Waksman ●スミ版 ●シアン版を紺色(前回と同色)に
2
色刷り
THE WAKSMAN FOUNDATION OF JAPAN INC.Published by
THE WAKSMAN FOUNDATION OF JAPAN INC.
THE WAKSMAN FOUNDATION
OF JAPAN INC.
Report of Researches in 2013
2 0 1 4
THE WAKSMAN FOUNDATION
OF JAPAN INC.
T H E W A K S M A N F O U N D A T IO N O F J A P A N IN C .
R e p o r t o f R e s e a r c h e s in 2 0 1 3 Dr. Selman A.Waksman ●スミ版 ●シアン版を紺色(前回と同色)に
2
色刷り
THE WAKSMAN FOUNDATION OF JAPAN INC.Published by
THE WAKSMAN FOUNDATION OF JAPAN INC.
THE WAKSMAN FOUNDATION
OF JAPAN INC.
Report of Researches in 2013
2 0 1 4
THE WAKSMAN FOUNDATION
OF JAPAN INC.
THE WAKSMAN FOUNDATION
OF JAPAN INC.
Honorary Patron
H.I.H. Prince Akishino
Board of Directors
Chairman : Ichiro Kitasato, Adviser, The Kitasato Institute.
Former Chairman of The Board, Meiji Seika Kaisha, Ltd.
Shogo Sasaki, Prof. Emeritus, Keio Univ.
Teruhiko Beppu, Prof. Emeritus, Tokyo Univ.
Takeshi Ishikawa, Vice President
Retirement Allowance Foundation of Private Colleges and Universities.
Tadakatsu Shimamura, Prof. Emeritus, Showa Univ.
Tadayoshi Shiba, Chairman Emeritus, The Kitasato Institute.
Managing
Director : Takeji Nishikawa, Prof. Emeritus, Keio Univ.
Comp-troller : Yoshiharu Wakiyama, Senior Adviser, Kaken Pharmaceutical, Co., Ltd.
Shirow Enoki, Former President, Seikagaku Corporation.
Councilors
Ryoichi Mori, Prof. Emeritus, Kyushu Univ.
Keizo Takemi, Member,The House of Councilors.
Koichi Yamanishi, Director General,The Research Foundation for Microbial Diseases of
Osaka Unoversity.
Sachiko Goto, Prof. Emeritus, Toho Univ.
Isao Uchida, Senior Adviser, Yokogawa Electric Corporation.
Shigeo Koyasu, D. Sc. Director RIKEN Center for Integrative Medical Sciences.
Takashi Shoda, Senior Corporate Adviser, Daiichi Sankyo Co., Ltd.
Yuko Kitagawa, Prof., Keio Univ. Sch. Med.
Special Adviser
Akira Uehara, Chairman & CEO, Taisho Pharmaceutical Holdings Co., Ltd.
Osamu Nagayama, Chairman & CEO, Chugai Pharmaceutical Co., Ltd.
Haruo Naito, President & CEO, Eisai Pharmaceutical Co., Ltd.
Seiichi Sato, President & CEO, Sato Pharmaceutical Co., Ltd.
Yoshihiro Miwa, President & CEO, Kowa Co., Ltd.
2 0 1
4
Edited and Published byTHE WAKSMAN FOUNDATION OF JAPAN INC. 30-8 Daikyo-cho, Shinjuku-ku,
Tokyo 160-0015, Japan http://www.waksman.or.jp/ E-mail: [email protected]
Printed by
D CRAFT SEIKOU CO., LTD. Tokyo, Japan
Preface to the First Report (1962)
It is indeed a privilege to take this opportunity to write a few words of introduction to the first report of the Waksman Foundation of Japan Inc., covering five years of its activities and comprising the results of the work of the first two years of research carried out by various scholars in Japan in the fields of microbiology and medical science, supported by this Foundation.
In 1952, I accepted the invitation from Keio University and the Kitasato Institute, to deliver the centennial lecture in honor of the great Japanese bacteriologist, Shibasaburo Kitasato. Before departing for Japan, I proposed to the trustees of the Rutgers Research and Educational Foundation which owned the patents on streptomycin, to share the royalties under the patent in Japan, for the support of research in microbiology and allied fields in that country. The trustees heartily approved my recommendation that I make such announcement to that effect.
Soon upon my arrival in Japan (December 17, 1952), I invited a group of eminent microbiologists, biochemists, and clinical investigators to meet with me in order to discuss the plan. Everyone present was very enthusiastic about the proposal. It was decided that a proper committee be selected to work out the plan of a Foundation under which the royalties were to be received and distributed for the support of Japanese investigators working in different universities in Japan and elsewhere, in the fields of microbiology and medical research. The committee recommended that a Board of Directors be selected and the proposed Foundation be named THE WAKSMAN FOUNDATION OF JAPAN INCORPORATION.
The Rutgers Research and Educational Foundation approved at once the above recommendations and issued a statement, signed by Dr. Lewis Webster Jones, President of the Foundation, to the effect that
‘‘The Rutgers Research and Educational Foundation desires to emphasize that its principal concern is the advancement of scientific knowledge in the public interest and that it confidently expects that the Waksman Foundation for Microbiology and Medical Research in Japan will be similarly motivated, thereby serving the peoples of both countries.’’
This announcement was received with enthusiasm both by the scientific world and the popular press in Japan and in the United States. It took several years before the Waksman Foundation of Japan Inc. was properly organized, and before applications were received and approved. In 1958, I had the privilege of participating in the first official meetings of the Board of Directors of the Japanese Foundation and to greet personally the first group of scholars to whom grants had been made.
In summarizing these brief remarks in connection with the first cinqueannual report of the Waksman Foundation of Japan Inc., I would like to enphasize that this example of collaboration between universities and scientists of the United States and Japan may serve to encourage collaboration between scientific workers throughout the world towards a better understanding between men and women and towards a happier and healthier human race, so that all the nations on this earth can live in peace and that man may finally ‘‘break
his swords and build out of them plowshares’’ for the betterment of mankind as a whole.
Selman A. Waksman Professor Emeritus
Rutgers-State University N. J., U. S. A.
The ‘‘Waksman Foundation of Japan Inc.’’ was established in 1957 with the spirit of humanity by Dr. S.A. Waksman, Professor of Microbiology, Rutgers University, U.S.A. The Foundation’s operations are possible only because Dr. S.A. Waksman and the Rutgers Research and Educational Foundation donated patent royalties he received from the production in Japan of the discovery, Streptomycin.
Because of these royalties, each year many Japanese scholars and research workers in the fields of Microbiology and medical science are encouraged and find it possible to continue their work. Especially, in accordance with Dr. Waksman’s suggestion, the funds are distributed to scholars in local and economically hampered schools and laboratories and to those developing research workers who are endeavoring to expand in their fields. This thought of Dr. Waksman’s is most appreciated, as it matches our Oriental phylosophy, and results in the search for a jewel among ordinary stones, which is the highest work of the science-leader.
Some five years have now passed since the start of this Foundation, and many persons have received aid through this period.
The reports which are presented herein cover the first and second group of research workers who received financial assistance from the Foundation.
Toshio Katow, M. D. Executive Director
Contents
Hirotaka Kanuka:
Genetic dissection of intermediate host and tapeworm interaction ……… 1
Jun-ichi Wachino:
Horizontal gene transfer of an amikacin resistant 16S rRNA methyltransferase
gene from multidrug resistant Acinetobacter baumannii ……… 7
Hayato Takahashi, M.D., Ph.D.:
Analysis on the mechanism of T cell peripheral tolerance against pemphigus
autoantigen, desmoglein 3 ……… 15
Hitomi Mimuro:
Helicobacter pylori regulate host non-coding RNA expression to increase
the proliferation of gastric epithelium during chronic infection ……… 23
── Report of Researches in 2013 ──
Genetic dissection of intermediate host and tapeworm
interaction
Hirotaka Kanuka
Department of Tropical Medicine, The Jikei University School of Medicine, Minato-ku, Tokyo 105-8461, Japan
ABSTRACT Dwarf tapeworm, Hymenolepis nana, which belongs to phylum Cyclophyllidea, is the most
com-mon cestode of human. Its intermediate host is arthropods, in particular, beetles. Once the intermediate host ingests tapeworm eggs, oncospheres immediately hatch and pass through insect gut wall. Cysticercoids develop within the hemocoel where they survive without loss of infectivity until the intermediate host is ingested by a definitive host. To dissect the interaction between tapeworm and intermediate host, we employed a reverse genetic approach with red flour beetle, Tribolium castaneum, in which a robust systemic RNA interference (RNAi) response is observed, as a model system to explore host responses to tapeworm infection. Adult knock-down phenotypes in T. castaneum were induced by injection of double-stranded RNA (dsRNA) into late instar larvae. We performed RNAi screening targeting several gene transcripts of Toll and immune deficiency (IMD) pathways, which are two major signal-ing pathways of humoral immune response in insects. Reduction of Toll pathway function, which was induced by RNAi-mediated silencing of MyD88, Dif1, and Dif2, in addition to JAK/STAT and JNK components, caused increase of burden of cysticercoids. On the other hand, RNAi-mediated knockdown of IMD pathway components, dredd and imd, had no significant difference on cysticercoid load. Our findings suggest a pivotal role of specific pathway such as Toll signaling in regulating resistance to tapeworm infection.
Introduction
Understanding the molecular machinery of the responses of disease-transmitting arthropods (vectors) against pathogens is of great importance for current efforts to develop novel strategies for control of vector-borne diseases. Arbovirus, protozoan parasites, and parasitic nematodes undergo substantial stage-specific losses during those developments in the vector mosquito using a certain defense system, which in some cases lead to complete refractoriness of the mosquito against those pathogens. On the other hand, susceptible mosquitoes have a greater tendency to hold sufficient number of parasites for transmission than refractory strains, acting as a “cargo of pathogen”. The ability of vector arthropods to possess human
pathogens (i.e., virus, protozoan parasites, and
worms) without showing any pathological symptoms is commonly referred to as the “vector paradox”, which remains the important and unanswered question concerning dual strategies of host (vector) defense: resistance and tolerance
Defense against pathogenic micro-organisms and other parasites can be divided into two conceptually
different components: resistance, a character that
reduces the pathogen’s opportunity of successful infection through an impact on pathogen fitness potentially via the host’s rate of pathogen clearance, and tolerance, the host’s ability to cope with the health impacts of a pathogenic encounter without a consequent reduction in fitness to the host. In vector arthropods, a distinguishing feature between these two strategies for dealing with pathogen spreading is that resistance has a negative effect on pathogens, whereas tolerance does not, as a result, their relative importance
in vector species may have substantial consequences for the evolution of vector-pathogen interactions. Solving and ultimately manipulating the vector paradox to control the vector-borne diseases is in many respects the “Holy Grail” for all of scientists in the research area. By continuing to unravel the mysteries that underlie the mutual prosperity between pathogen and vector, we believe that it may one day be possible to selectively attenuate the tolerance activities of vectors in addition to enhance the resistance and, consequently, to “minimize” vector fitness/health in such a way that vector itself is converted from a life-threatening arthropods to one that is a fragile blood-sucking bug with a weak constitution unable to endure the pathogen transmission.
Human tapeworms, such as Hymenolepis nana, require insect as intermediate host to infect definitive
host. The tapeworm, which belongs to phylum
Cyclophyllidea, is the most common cestode of human. Its intermediate hosts are grain pest beetles and cockroaches. Humans get infected with tapeworm by accidental ingestion of infected insects. When the intermediate host insect ingests tapeworm eggs, oncospheres immediately hatch and pass through insect gut wall. Cysticercoids develop in the hemocoel where they survive without loss of infectivity until the intermediate host is ingested by a definitive host. For this reason, it has been supposed that the response of intermediate host insects to tapeworm is absent or weak.
Here we focus to determine the signal transduction pathways for tolerance, by using insect genetic system, and more importantly, to identify and characterize the manner through which artificial aberrations in the tolerance process contribute to the disruption of pathogen spreading by vectors. To dissect the tolerance in addition to resistance system, we employed a reverse genetic approach with red flour beetle, Tribolium castaneum, in which a robust systemic RNA interference (RNAi) response is observed, as a model system to explore host responses to pathogens infection. Our approach with specific aims designed will contribute to uncover common features in the tolerance system that may have in tern contributed to the certain specific characteristic of vector species.
Materials and Methods Beetle Cultures
The beetle cultures were reared on whole wheat
flour [+5% (wt/wt) yeast] at 30◦C in a temperature and
humidity controlled incubator. An enhancer trap line pu11, which has enhanced yellow fluorescent protein (EGFP) expression in the hindwing and elytron discs, was used for all RNAi experiments.
Tapeworm Parasite
Hymenolepis nana used in this study is parasitic tapeworm of human and rodent. The tapeworm is
relatively small (∼ 4 cm) and has short life cycle (∼ 1
month). The tapeworm does not need intermediate host to complete its experimental life cycle because of direct ingestion of tapeworm eggs. The passage of this tapeworm is accomplished by using mice as experimental host. H. nana (a gift from Dr. Kazuhito Asano, Showa University) has been maintained in laboratory for more than 20 years. Preparation of H. nana eggs and oral infection of mice were performed as previously described (Asano and Muramatsu, Int J Parasitol 27(11): 1437-43 (1997)). Briefly, mice were challenged orally with about 1000 H. nana eggs and killed 4 days later. All parasites derived from the primary infection were recovered as lumen-dwelling adult worms.
Gene Cloning and dsRNA Synthesis
The Tribolium ortholog of Drosophila gene was identified via BLAST analysis. Tribolium pupal cDNA was used to clone the cDNA fragment into pT7 vector (Invitrogen). The dsRNA templates were synthesized by PCR using the vector primer, or gene specific primers with the T7 polymerase promoter sequence at the 5’ end. dsRNAs were synthesized by in vitro
transcription (Megascript T7; Ambion) using 1.5μg
of templates. The resulting dsRNA samples were then purified by Megaclear kit (Ambion). Specificity of the products was confirmed via agarose gel electrophoresis.
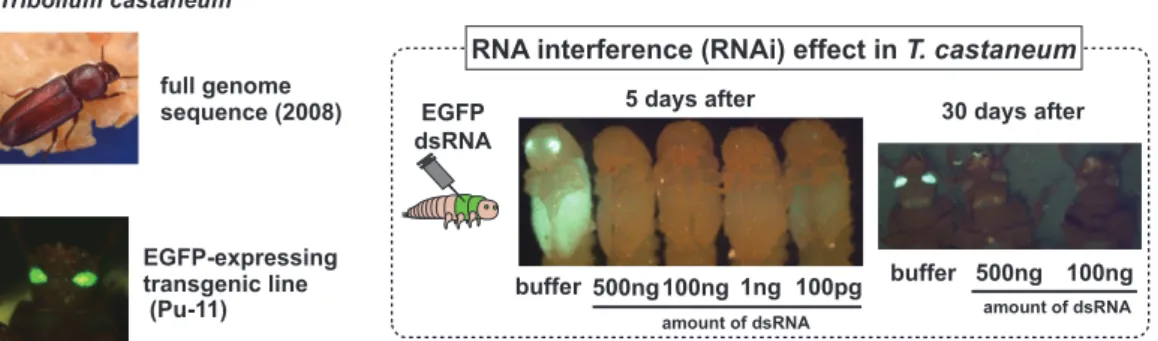
EGFP-expressing transgenic line (Pu-11) full genome sequence (2008) Tribolium castaneum amount of dsRNA 5 days after 30 days after buffer 500ng 100ng 1ng 100pg buffer 500ng 100ng EGFP dsRNA
RNA interference (RNAi) effect in T. castaneum
amount of dsRNA
Figure 1. Tribolium castaneum exhibits a robust systemic RNA interference response.
pu11 larvae, pupae, and adults after injection with buffer early in the last larval stage. Green fluorescent protein (GFP) expression in these individuals is identical to non-injected pu11 beetles. pu11 larvae injected with various concentrations of GFP dsRNA. Injection of 100 pg dsRNA in each larva induces almost perfect silencing in most larvae
Concentration of dsRNA ranged from 5 to 7μg/uL.
dsRNA Injection into Beetle
Injections were performed in the last larval stage or penultimate larval stage. EGFP expression in pu11 developing wing and elytron discs was used to select appropriate larval stages. Stereomicroscopes (Leica) were used for the injections. At least 50 larvae were used for each set of injections. Approximately 0.7–
0.9μL of 1 μg/μL dsRNA solution was injected into
each larva. After injection, the larvae were kept in
flour at 30◦C until they became adults for phenotypic
analysis.
Parasite Infection into Beetle
Adult loss-of-function phenotypes were examined by injection of 100 ng dsRNA into late instar larvae or penultimate larvae of pu11 beetles expressing EGFP as described above. After 3 days starvation, artificial infection was performed by direct feeding of adult tapeworms to the beetle, which was collected from small intestine of infected mice. Twelve intact tapeworms were fed to thirty beetles. After 10 days from infection, beetles were dissected to quantify the number of cysticercoid.
Results
To investigate signaling regulation interaction between parasitic worm and intermediate host, we performed a loss-of-function (RNAi-based) screening to find genes that affect tolerance and/or resistance in beetles infected with tapeworm. No observable decrease in tapeworm load in prolonged survival strains against infection will be taken as an indication of a shift in the balance between resistance and tolerance of host defense to a state of coexistence with the invading parasite.
To dissect interaction between tapeworm and intermediate host, we employed RNAi screening with T. castaneum, as a model system to explore host responses to tapeworm infection. The RNAi effect can be elicited in any tissue and any stage by the injection of dsRNA into the hemocoel, and injection of dsRNA into adult females can even be used to identify phenotypes in offspring. In order to confirm RNAi effect in T. castaneum, dsRNA for EGFP was injected to larvae of pu11 beetles (EGFP expressing enhancer trap line). The silencing effect was observed even at 100 pg/larva and sustained for 30 days (Figure 1).
Transcriptomes of beetle midgut, which is
crucial interface between beetles and tapeworms, were analyzed using next-generation sequencing (Figure 2). This transcriptome analysis resulted in the identification of 203 differentially expressed genes
Figure 2. Tribolium castaneum infection increases expression of immune-related genes.
Transcriptomes of T. castaneum midgut, which is crucial interface between beetles and tapeworms, were analyzed using next-generation sequencing. Pie chart represents the classification of 203 differentially expressed genes with statistical significance based on GO biological process.
0 10 20 30 40 50 Dif2 EGFP <0.05 0 10 20 30 40 Dif1 EGFP p<0.05 0 10 20 30 40 50 60 <0.01 MyD88 EGFP 0 20 40 60 0 12 140 ≈ spätzle1 EGFP <0.01 p p p
Figure 3. Toll pathway is involved in resistance to tapeworm infection in Tribolium castaneum.
Reduction of Toll pathway function, which was induced by RNAi-mediated silencing of MyD88, Dif1, Dif2 and Sp¨atzle1, caused increase of burden of cysticercoids. For scatter plot, the each dot represents cysticercoid counts from individual beetles and the horizontal black bar indicates the median infection level. dsRNA for EGFP-injected groups were used as negative control.
upon tapeworm infection. Furthermore, tapeworm infection caused elevated expression of 5 genes encoding antibacterial peptides. This result suggests that tapeworm infection induces innate immunity generating antibacterial peptides. We then selected 101 genes for primary RNAi screening, not only innate immunity-related genes but also Tribolium ortholog of host defense-related genes (e.g., JNK and JAK/STAT genes), which were previously reported in other arthropods.
Reduction of Toll pathway function, which was induced by RNAi-mediated silencing of MyD88, Dif1, Dif2 and Sp¨atzle1, caused increase of burden of
cysticercoids (Figure 3). These results suggest that innate immunity induced by Toll pathway signaling suppresses tapeworm infection. The contribution of Toll pathway to parasite infection was already reported in mosquito-plasmodium studies, thus these results confirm the validity of this RNAi screening method. On the other hand, RNAi-mediated knockdown of IMD pathway components, dredd and imd, had no significant difference on cysticercoid load (data not shown).
RNAi-mediated depletion of JAK/STAT pathway components, Domeless (encoding a receptor for a ligand stimulating JAK/STAT) and Hopscotch (JAK kinase), increased cysticercoid burden. On the other
0 50 100 150 Hopscotch EGFP p<0.01 0 20 40 60 80 SOCS36E EGFP p<0.01 0 50 100 150 Domeless EGFP p<0.01
Figure 4. JAK/STAT pathway is involved in resistance to tapeworm infection in Tribolium castaneum.
RNAi-mediated depletion of JAK/STAT pathway components, Domeless and Hopscotch, increased cysticercoid burden. The silencing of negative regulator of this pathway, SOCS36E, reduced cysticercoid load. For scatter plot, the each dot represents cysticercoid counts from individual beetles and the horizontal black bar indicates the median infection level. dsRNA for EGFP-injected groups were used as negative control.
0 20 40 60 80 basket1 p<0.01 EGFP 0 20 40 60 80 p<0.05 basket2 EGFP 0 20 40 60 80 slipper EGFP p<0.01
Figure 5. JNK pathway regulates susceptibility to tapeworm infection in Tribolium castaneum.
Knock-down of the JNK pathway components, basket1, basket2 and slipper, enhanced tapeworm infection. For scatter plot, the each dot represents cysticercoid counts from individual beetles and the horizontal black bar indicates the median infection level. dsRNA for EGFP-injected groups were used as negative control.
hand, silencing of negative regulator of this pathway, SOCS36E, reduced cysticercoid load (Figure 4). These results suggest that JAK/STAT pathway facilitates tapeworm infection. We also found that deletion of the JNK pathway components, basket1, basket2 (encoding JNK, respectively), and slipper (encoding upstream kinase of JNK pathway), enhanced tapeworm infection (Figure 5), suggesting that the host response induced by JNK pathway signaling reduces tapeworm burden. Eiger, the TNF-superfamily ligand in T. castaneum, induces cell death through the activation of JNK signaling. Eiger plays a central role in cell competition, which is a mechanism to keep tissue homeostasis. RNAi-mediated silencing of Tribolium Eiger gene caused increase of the number of cysticercoid (Figure
6). These results suggest that JNK pathway-induced stress response, which is independent of innate immunity, limits tapeworm infection.
To get deeper mechanistic insights about tolerant mechanism in insects and to apply these findings into vector species, it is necessary to describe the conserved tolerance system via analyzing the knock-down phenotypes of candidate tolerance-related genes in particular p38 MAP kinase family in Tribolium in addition to the fly (Shinzawa et al., Cell Host Microbe 6(3): 244-52 (2009)). Elucidating the mechanisms by which vector-specific traits arose may shed light on one of the general principles of host tolerance diversity. However, we determined the p38 loss-of-function phenotypes in Tribolium and observed significant effect
0 10 20 30 40 50 60 70 p<0.01 th e number of cysticercoid Eiger EGFP
Figure 6. TNF ligand is involved in defense response to tapeworm infection in Tribolium castaneum. Eiger, the TNF-superfamily ligand in T. castaneum, induces cell death through the RNAi-mediated silencing of T. castaneum Eiger gene encoding a TNF-superfamily ligand caused increase of the number of cysticercoid. The each dot represents cysticercoid counts from individual beetles and the horizontal black bar indicates the median infection level. dsRNA for EGFP-injected groups were used as negative control.
on neither cysticercoid burden nor survival rate of host beetle (data not shown), suggesting that there could be alternative pathways controlling host tolerance against tapeworm infection.
Our findings in this report indicate that JAK/STAT and JNK pathways play pivotal role to regulate resistance to tapeworm infection in addition to Toll-mediate innate immunity. It is supposed that the integration of several host responses may be important to define the proper competency of intermediate host for tapeworm transmission.
Discussion
Effective control strategies of parasitic diseases targeting intermediate host currently include insecticide treatment delivered through spraying of houses or insecticide-impregnated insect nets. While these methods are effective at decreasing arthropod numbers, they also contribute to the rise of insecticide-resistance intermediate host species. An alternative strategy involves inhibition of parasite development within the intermediate host. One such strategy involves release of transgenic intermediate host resistant to parasite that could compete with wild populations.
However, transgenes have proven difficult to fix in populations. The finding that Toll and JAK/STAT pathways in immune response tissues was capable of conferring resistance to tapeworm H. nana was surprising considering traditional roles for the Toll and JAK/STAT pathways; the JAK/STAT pathway has been thought to be activated in response to virus while the Toll pathway was responsible for immunity to Gram-positive bacteria and fungi. Recently this view of insect innate immunity has begun to change with numerous findings contradicting traditional views. Certainly, this study provides further evidence for a modified view of innate immunity against large parasite such as tapeworm.
While this study clearly demonstrates a novel role for JNK and JAK/STAT pathways in conferring resistance to beetles infected with tapeworm, a role for these factors in resistance cannot be ruled out. Indeed, survival of an infected organism likely depends on a combination of both resistance to that organism and tolerance to the infection. Such a combination of mechanisms can lead to a total of 9 different states of an organism ranging from high tolerance coupled with low resistance to low tolerance coupled with high resistance and all states in between. The JNK and JAK/STAT-mediated defense seen in this study potentially falls into the class represented by high resistance and low tolerance. In order to shed light on such complicated defense mechanisms comprehensive work investigating the synergistic effects of both tolerance and resistance to host fitness will be required.
Conclusion
Invertebrate model systems such as Tribolium are recognized as a powerful means of studying conserved mechanisms and genes relevant to human health. In this research proposal, through bridging from Tribolium to vector species, our work will greatly extend the knowledge of the molecular basis of tolerance in pathogen transmitting vectors. Our findings will offer important insights and inroads in understanding this important and essential question that clearly underlies the biological and pathological actions of vector tolerance.
Horizontal gene transfer of an amikacin resistant 16S
rRNA methyltransferase gene from multidrug resistant
Acinetobacter baumannii
Jun-ichi Wachino
Department of Bacteriology, Nagoya University Graduate School of Medicine, 65 Tsurumaicho, Showa-ku, Nagoya, Aichi 466-8550, Japan
Introduction
Acinetobacter baumannii is an opportunistic Gram-negative pathogen that causes severe infections such as ventilator-associated pneumonia in hospitalized patients and is frequently associated with nosocomial outbreaks (1). A. baumannii is becoming a serious threat to public health due to the acquisition of multidrug-resistant genes. This has resulted in the emergence of A. baumannii strains resistant to carbapenems, aminoglycosides, and fluoroquinolones, which limits the choice of antimicrobial agents for the treatment of clinical infections. The antimicrobials tigecycline and colistin are used as a last resort in the treatment of multidrug-resistant (MDR) A. baumannii. However, even tigecycline and colistin resistant strains have recently emerged (2, 3). The rapid spread of MDR A. baumannii has been attributed to the transmission of several international strains, which are assigned to International clones (IC) I and II (4). Of these, the
IC-II is emerging as the world's dominant population of
MDR A. baumannii.
One of the aminoglycoside resistance mechanisms identified in MDR A. baumannii is the aminoglycoside resistance 16S rRNA methyltransferase gene (armA), which prevents aminoglycoside from binding to the ribosome through modification within the aminoacyl-tRNA binding site of the 16S rRNA (5). Aminoglyco-side resistance has been widely disseminated through the acquisition of armA among A. baumannii isolates.
Galimand et al. has shown that the translocation of armA associated with Tn1548 could occur from a non-conjugative plasmid of a Klebsiella pneumoniae clini-cal isolate using a geneticlini-cally tractable Escherichia coli system (6). It is therefore possible that the translocation of the armA gene could also occur in A. baumannii as seen in K. pneumoniae.
Recently, the complete genome sequences for a number of endemic MDR A. baumannii belonging to the IC-II have become available. Genome analysis revealed that the armA gene is located within the chromosome for the majority of the IC-II strains. We hypothesized that the IC-II is a major source for the distribution of the armA gene to other bacterial species, including antibiotic sensitive non-baumannii Acinetobacter species. Acinetobacter species are naturally transformable (7), an attribute which is thought to accelerate the dissemination of antibiotic resistance genes. However, the contribution of this feature to the emergence of MDR A. baumannii has yet to be determined. The aim of this study was to elucidate the molecular mechanisms behind the transfer of antibiotic resistance genes, focusing on the aminoglycoside resistance gene armA, in A. baumannii. Materials and methods
Bacterial strains and growth conditions
The A. baumannii clinical strain ARS60, which was isolated from a patient sputum sample in 2004,
was used in this study. The antibiotic susceptible A. baumannii laboratory strain ATCC17978 was used as the recipient strain. The bacterial strains were grown in Luria-Bertani (LB) agar and broth, with the addition of
amikacin (25μg/ml) when required.
Genome sequencing of A. baumannii strains
Genomic DNA was extracted from the A. baumannii strains using a QIAamp DNA Mini kit. The genomic DNA was sheared to a size of approximately 500 bp, performed by Covaris. The sheared genomic DNA was purified using a MinElute PCR purification kit. Pyrosequencing was performed on a 454 GS junior. The A. baumannii ARS60 strain reads were assembled into contigs by the de novo assembler (Newbler Assembler version 2.3). The A. baumannii ATCC17978ΩarmA reads were first mapped to the reference genome of the A. baumannii ATCC17978 strain using a reference mapper program. The remaining unmapped reads were further assembled into contigs by the de novo assembler. This allowed the identification of foreign DNA inserted into the genome of A. baumannii ATCC17978.
Transposon detection using PCR
The presence of a circular intermediate (CI) was detected by inverse PCR, using the primer sets, P1
(5'-ccatcacatgtatgaccaga-3') and P2 (5'
-gtttttccagtac-tacgcca-3'). Evidence of genetic rejoining following
ex-cision of a composite transposon was confirmed by
PCR, using the primer sets P3 (5'-aattgggctgatgagtacgg
-3') and P4 (5'-gctgcacagtggttttctga-3').
Susceptibility testing
The minimum inhibitory concentrations (MICs) were determined. Bacteria samples were adjusted to
108cfu/ml, and spread on a Mueller-Hinton agar plate.
The Etest strip was placed onto the plate and incubated
at 35◦C for 18 h.
Purification of the component responsible for the transfer of armA
The A. baumannii ARS60 strain was cultured in LB broth and the bacterial cells were removed by centrifugation. The supernatant was passed through
0.45μm filter and ultracentrifugation was carried
out at 150,000 × g for 90 min at 4◦C. After
ultracentrifugation, the pellets were resuspended with phosphate buffered saline (PBS), passed through
0.22μm filter and stored at −80◦C until required.
Transformation
The purified component from the bacterial super-natant was combined with A. baumannii ATCC17978 and incubated on LB agar plates. After incubation, the bacteria growth was scraped off with a sterile loop, re-suspended in LB broth and spread onto LB LB agar
plates supplemented with 25μg/ml amikacin.
Results
Antibiotic resistance genes
The draft genome sequence of the A. baumannii ARS60 strain consisted of 144 contigs, corresponding
to 3,963,199 bp, with an N50 contig size of 60,507
bp. The web-based tool, ResFinder version 1.3 was used to interrogate the draft genome sequence, in order to identify acquired antibiotic resistance genes. The analysis showed that several antibiotic resistance genes were embedded within mobile genetic elements, located on the chromosome of the ARS60 strain (Table 1). Due to the acquisition of these antibiotic resistance genes, A. baumannii ARS60 has become a multidrug resistant strain. The A. baumannii ARS60 strain has been assigned to ST208 by the Bartual scheme, ST2 by Pastuer Institute Scheme and belongs to the IC-II. Genetic environment of armA
The genetic platform containing the armA gene, located on the chromosome of the A. baumannii ARS60
Table 1. Antibiotic resistance genes found in A. baumannii ARS60 strain
Resistance gene Function Resistance Phenotype
aac(3)-Ia acetyltransferase aminoglycoside
aadA1 adenylyltransferase aminoglycoside
aac(6')-Ib acetyltransferase aminoglycoside
aph(3')-Ic phosphotransferase aminoglycoside
strA phosphotransferase aminoglycoside
strB phosphotransferase aminoglycoside
armA 16S rRNA methyltransferase aminoglycoside
blaOXA−66 β-lactamase β-lactam
blaADC−25 β-lactamase β-lactam
blaTEM−1D β-lactamase β-lactam
mph phosphotransferase macrolide
mel efflux transporter macrolide
catB3 acetyltransferase chloramphenicol
sul1 dihydropteroate synthase sulfonamide
tetB efflux transporter tetracycline
strain (Fig. 1), was revealed following analysis of the draft genome sequence and the use of the PCR to close the gaps between contigs. Upstream of the armA gene lies a putative transposase gene, ISCR1 (previously called orf513). The classical class 1 integron includes
ΔintI1 at its 5'-conserved segment (CS), the antibiotic
resistance gene cassettes aac(6')-Ib, catB3, aadA and
qacEΔ1-sul1 at its 3'-CS. Downstream of the armA
gene, are macrolide resistance genes mel and mph, a hypothetical protein, IS66, and a putative rep gene. This 18.3 kb region containing the armA gene was flanked by two IS6 elements, with 14-bp inverted repeats (IRL and IRR) at the terminal ends, indicating a composite transposable element. The same 8 bp nucleotide sequences, CTCATCCT was located at both terminal sides of the composite transposable element. The presence of these 8 bp target sites would be an indication that insertion of the transposon bearing the armA gene had occurred at this position.
Detection of circular transposon DNA and postexcision site
It is likely that the chromosomally located 18.3 kb composite transposable element containing the armA gene could form a non-replicative CI, before lateral
transposon transfer. To confirm the CI formation, inverse PCR was performed using the genomic DNA extracted from the A. baumannii ARS60 strain. If the composite transposon excises and forms a CI, a PCR product would be generated. The 1.2 kb PCR product generated by the P1 and P2 primer sets, confirmed the presence of the CI (Fig. 2). In addition, we hypothesized that there would be evidence for the gene rejoining following the excision of transposon. To test this we used PCR and confirmed the presence of a 1.5 kb fragment produced by P3 and P4 primer sets, indicating the left IS6 element (Fig. 2). These results suggest that CI formation occurred among a small proportion of A. baumannii ARS60 population
Transfer of the armA gene in vitro
Components purified from the A. baumannii ARS60 strain supernatant by ultracentrifugation, were combined with the A. baumannii ATCC17978 strain. After incubation, the ATCC17978 transformants that had acquired amikacin resistance were selected on LB agar plates containing amikacin, as shown in Fig. 3. The acquisition of amikacin resistance was demonstrated when the components purified from the A. baumannii ARS60 supernatant following 6 h of growth, were
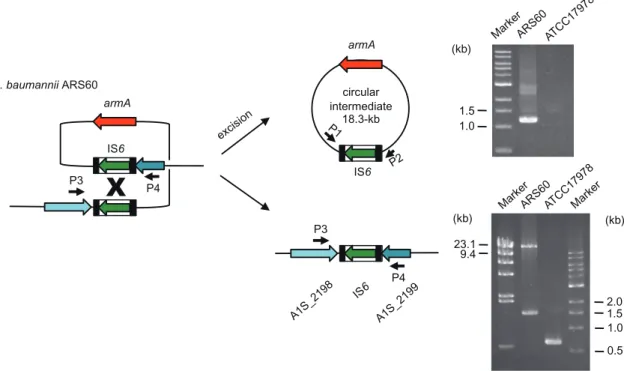
A1S_2192 A1S_2193 A1S_2194 A1S_2196 A1S_2197 A1S_2198 A1S_2199 A1S_2200 A1S_2201 A1S_2202 A1S_2203 A1S_2204 A1S_2205 A1S_2206 A1S_2207 A1S_2208 A1S_2209 A1S_2210 A1S_2211 A1S_2212 A1S_2213 A1S_2214 A1S_2215 60_2192 60_2193 60_2194 60_2196 60_2197 60_2199 60_2200 60_2201 60_2202 60_2203 60_2204 60_2205 60_2206 60_2207 60_2208 60_a 60_b 60_2198 IS6 rep IS66 mph mel hyp armA tmpAcp1 ISCR1 sul1 qacE 1 aadA1 catB3 intI1 IS6 19-kb (composite transposon) 4.3-kb 60_2212 60_2213 60_2214 60_2215 60_2200 60_2201 60_2202 60_2203 60_2204 60_2205 60_2206 60_2207 60_2208 A1S_2192 A1S_2193 A1S_2194 A1S_2196 A1S_2197 A1S_2198 1-kb A TCC17978 ARS60 17978 armA A1S_2209 A1S_2210 A1S_2211 A1S_2212 A1S_2213 A1S_2214 A1S_2215 60_2199
Fig. 1. Comparison of the region surrounding the armA gene in A. baumannii ARS60 and A. baumannii ATCC17978ΩarmA. The genes for A. baumannii ATCC17978 are shown with white arrows and those for A. baumannii ARS60 are shown with light green arrows.
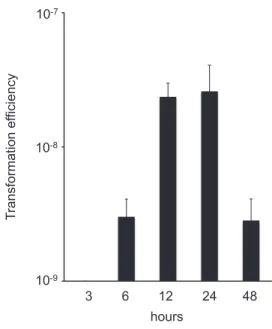
mixed with the A. baumannii ATCC17978 strain, and increase to 24 h, and then decrease to 48 h. When the A. baumannii ATCC17978 strain was not combined with the purified components from the A. baumannii ARS60 strain supernatant, resistant colony did not appear. Several colonies which had grown on agar plates supplemented with amikacin were subjected to PCR, and the presence of the armA
gene was confirmed. PFGE and Southern hybridization revealed that the armA gene was integrated into A. baumannii ATCC17978 chromosome (data not shown).
The transformants (named 17978ΩarmA) showed the
same high levels of amikacin resistance as the ARS60 parent strain (Fig. 4).
circular intermediate 18.3-kb IS6 armA A. baumannii ARS60 1.5 1.0 (kb)
X
23.1 9.4 2.0 1.5 1.0 0.5 (kb) (kb) P3 armA IS6 P4 P3 P4Fig. 2. Detection of circular intermediate carrying the armA gene and the site following excision.
Fig. 3. Results of amikacin susceptibility testing by the Etest for A. baumannii ARS60 strain, A. baumannii ATCC17978ΩarmA, and A. baumannii ATCC17978.
10-9 10-8 10-7 T ransformation ef ficiency 3 6 12 24 48 hours
Fig. 4. Transformation efficiency for A. baumannii ATCC17978 following transformation with pu-rified components from A. baumannii ARS60 supernatant. Error bars indicate the standard deviation.
Acquisition of the ARS60 strain DNA by the transformant 17978ΩarmA
The genome sequence of the transformant
17978ΩarmA was determined by NGS analyses and
the results are shown in Fig. 1. The transformant
17978ΩarmA acquired a DNA fragment approximately
34 kb in size, which contained a composite transposon carrying the armA gene derived from the A. baumannii ARS60 strain.
Discussion
A. baumannii isolated from clinical settings have rapidly acquired resistance to a number of
antimicrobials, through the uptake of antibiotic
resistance genes. It is assumed that horizontal gene transfer (HGT) plays a crucial role in acquiring
antibiotic resistance genes by this organism, as
Acinetobacter species have an inherent ability to efficiently acquire exogenous DNA. The A. baumannii clinical isolate was also expected to take up antibiotic resistance genes through HGT; however, the details have not yet been fully elucidated. We hypothesized
that A. baumannii has a specific system to efficiently deliver resistance genes in order to bestow resistance to antibiotic sensitive Acinetobacter species.
In this study, we have succeeded in purifying components, which include DNA fragments encoding antibiotic resistance genes, from the culture medium. We have used these purified components to transform an antibiotic sensitive laboratory strain, and gener-ated aminoglycoside resistant bacteria. Rumbo et al reported that MDR A. baumannii clinical isolates re-leased outer membrane vesicle (OMV), which con-tained small plasmids bearing carbapenem resistance genes. These OMVs played a crucial role in transfer-ring antibiotic resistance genes to other A. baumannii strains (8). The armA gene of the A. baumannii ARS60 strain was carried by a composite transposon, which could be circularized (Fig. 2). It was speculated that cir-cularized transposons carrying the armA gene might be presented in the OMV delivered to other A. baumannii strains as suggested by Rumbo et al (8). We confirmed that the CI carried the armA gene in the ARS60 strain. However, the aminoglycoside resistant transformant,
17978ΩarmA acquired a DNA fragment approximately
34 kb larger than the composite transposon carrying the armA gene, probably through a homologous recombi-nation event. Therefore, the CI carrying the armA gene does not appear to be directly involved in the dissemi-nation of the armA gene, at least under the experimental conditions used in this study.
Our results indicate the presence of another mech-anism by which HGT is achieved in A. baumannii. We were able to further purify the supernatant components, which included DNA fragments, using sucrose density gradient centrifugation and gel filtration chromatog-raphy, and succeeded in transferring the armA gene in vitro. Purified components were resistant to DNase treatment, suggesting that the DNA fragments may be protected by OMV. Our future research aims include uncovering the mechanism responsible for the HGT of antibiotic resistance genes in A. baumannii.
References
(1) Howard, A., O'Donoghue, M., Feeney, A., et
opportunistic pathogen. Virulence 3: 243-250. (2) Vila, J., and Pachon, J. 2012. Therapeutic
options for Acinetobacter baumannii infections: an update. Expert Opin Pharmacother 13: 2319-2336.
(3) Sun, Y., Cai, Y., Liu, X., et al. 2013. The emergence of clinical resistance to tigecycline. Int J Antimicrob Agents 41: 110-116.
(4) Zarrilli, R., Pournaras, S., Giannouli, M., et al. 2013. Global evolution of multidrug-resistant Acinetobacter baumannii clonal lineages. Int J Antimicrob Agents 41: 11-19.
(5) Wachino, J., and Arakawa, Y. 2012. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: an update. Drug Resist Updat
15: 133-148.
(6) Galimand, M., Sabtcheva, S., Courvalin, P., et al. 2005. Worldwide disseminated armA amino-glycoside resistance methylase gene is borne by composite transposon Tn1548. Antimicrob Agents Chemother 49: 2949-2953.
(7) Overballe-Petersen, S., Harms, K., Orlando, L. A., et al. 2013. Bacterial natural transformation by highly fragmented and damaged DNA. Proc Natl Acad Sci USA 110: 19860-19865.
(8) Rumbo, C., Fernandez-Moreira, E., Merino, M., et al. 2011. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: a new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob Agents Chemother 55: 3084-3090.
Analysis on the mechanism of T cell peripheral
toler-ance against pemphigus autoantigen, desmoglein 3
Hayato Takahashi, M.D., Ph.D.
Department of Dermatology, Keio University School of Medicine, Shinjuku, Tokyo 160-8582, Japan
Introduction
Pemphigus vulgaris (PV) is a life-threatening bullous disease. In patients with PV, anti-desmoglein 3 (Dsg3) IgG autoantibodies are detected in the serum (1). Dsg3 is a cadherin-type transmembrane glycoprotein that is expressed in keratinocyte and critical in cell-cell adhesion of the cells. The autoantibodies bind keratinocyte cell surfaces in vivo and disturb cell adhesion function of Dsg3, resulting in acantholysis that is loss of cohesion between keratinocytes characteristically seen in PV histology. Since Dsg3 is expressed not only in the skin but also mucosal membrane including oral mucosa and esophagus and so on, anti-Dsg3 antibodies can induce erosion and blister not only in the oral mucosa and esophagus but also in the skin in combination with anti-Dsg1 autoantibodies as clinical manifestations (2).
Some previous reports support that autoreactive T cells are involved in and regulate the anti-Dsg3 antibody production from autoreactive B cells. For example, nucleotide sequencing of anti-Dsg3 antibody gene isolated from PV patients detected somatic hyper-mutations in CDR3 region of the antibody, revealing T cell-dependent affinity maturation of autoantibodies (3). PV is associated with HLA-DR14/DQB1*0503 in Japanese and HLA-DRB1*0402
in Jewish, suggesting that CD4+T cells recognize Dsg3
peptide presented in the MHC class II molecules (4-6). These results indicate that Dsg3-reactive T cells play a critical role in PV pathogenesis and can be a therapeutic target to control the disease.
Previously PV mouse model is generated by
trans-fer of lymphocytes from Dsg3-/- mouse into Rag2
-/-mouse (7). In Dsg3-/- mouse, tolerance mechanism
against Dsg3 is not established because of absence of Dsg3. Therefore, transferred lymphocyte can efficiently start to respond to Dsg3 and induce anti-Dsg3 antibody
production and PV phenotype in Rag2-/- mouse that
physiologically expresses Dsg3. Recently we
estab-lished Dsg3-reactive T cell clones from Dsg3-/-mouse
and demonstrated the pathogenicity of the clones by confirming PV phenotype induction after transferring
the clones with Dsg3-/-B cells into Rag2-/- mouse (8).
Furthermore, blocking IL-4, which is detected as a sig-nificantly up-regulated cytokine in pathogenic clones but not in non-pathogenic ones, ameliorates PV pheno-type. These results clearly support that Dsg3-reactive T cells is critical in PV mouse model.
While biologics, in addition to conventional drug such as steroid and immunosuppressant, are newly introduced for treatment of autoimmune diseases, antigen-specific therapy that harbors much less side effect is not clinically available yet. The purpose of this study is to understand molecular mechanism of immunological tolerance against Dsg3 and provide the basis to invent new therapeutic approach targeting autoreactive T cells. Especially it would be more important to understand peripheral tolerance but not central one since peripheral tolerance is more approachable than thymus when considering practical treatment in patients. In this study, we generated and analyzed Dsg3-specific T cell receptor transgenic mouse to understand behavior of Dsg3-specific T cells in immunological tolerance.
Materials and Methods Mice
C57BL/6 mice (H-2b, Ly-9.2) and 129/SV
(H-2b, Ly-9.1) were purchased from CLEA Japan (Tokyo,
Japan) and Sankyo Labo Service Corporation (Tokyo,
Japan). C57BL/6 Rag-2-/- mice were purchased from
Central Institute for Experimental Animals (Tokyo,
Japan). Dsg3-/-mice with a mixed genetic background
of 129/SV and C57BL/6J, and 129/SV Dsg3-/- mice
were obtained by mating male and female Dsg3
-/-mice (Jackson Laboratory, Bar Harbor, ME) (7).
Dsg1tg/tg-Dsg3-/-mice with C57BL/6 background were
generated as described in the other article (9). Animals were housed under specific pathogen-free conditions. The Keio University Ethics Committee for Animal Experiments approved all experiments in this study. Transgenic vector
Genes for TCR α and β chains of pathogenic
Dsg3-reactive T cell clone, 140#27, were subcloned
into transgenic expression cassettes for TCR α and
β chains, which were kindly provided by Dr. Diane Mathis (Harvard University) (10) following appropriate treatment with restriction enzymes.
Generation of Dsg3-reactive TCR transgenic mice
Linearized transgenes of TCRα and β chains for
a T cell clone, 140#27, were injected into fertilized
oocyte of C57BL/6J (H-2b) mice to generate TCR
transgenic mice, C57BL/6J-Tg(Dsg3TCR140), which were maintained by mating C57BL/6J mice.
Peptide
111 overlapping 15-mer peptides covering EC1 to EC3 of Dsg3 extracellular domain (1 - 394 aa) were synthesized and purchased from Sigma-Aldrich Japan (Tokyo, Japan).
In vitro reconstitution of Dsg3-reactive TCR
T cell hybridoma, TG40-CD4, which was kindly provided by Dr. Takashi Saito (Riken Research Cen-ter for Allergy and Immunology), looses intrinsic TCR expression and expresses mouse CD4, which
was retrovirally introduced. 10μg linearized
vec-tors for TCR α and β chain expression in
addi-tion to pSV2-hph (ATCC, Rockville, MD) contain-ing hygromycin-resistance gene were electrically intro-duced into TG40-CD4. Stable transfectants were se-lected by using hygromycin after electroporation. Ex-pression of transduced vectors was confirmed by flow
cytometry detecting co-expression of TCRβ and CD3,
which is expressed as a complex with both TCRα and
β chains. Stable transfectants were sorted by MACS
cell isolation system using anti-TCRβ6 Ab-PE (Becton
Dickinson, San Diego, CA) in combination with anti-PE Ab-microbeads (Miltenyi Biotech, Bergisch Glad-bach, Germany).
Reactivity of T cells and T cell hybridomas against Dsg3 peptides
Single cell suspension of 4× 104 cells were
prepared from the spleen of TCR transgenic mice and
cultured with 1× 10540 Gy-irradiated splenocytes and
10μg/ml peptide in 96-well round bottom plate and
Dsg3-specific T cell proliferation was measured by3
H-thymidine uptake as previously described (11). 2× 104
T cell hybridoma cells were cultured with 1× 106
40 Gy-irradiated splenocytes and peptide of indicated concentration in 96-well flat bottom plate for 24 hours. Culture supernatants were subsequently subjected to IL-2 ELISA (BD). Some of the experiments were performed in combination with anti-MHC class II mAb (M5/114) or isotype-matched rat mAb (BD).
Flow cytometry
Single cell suspension of thymus, spleen, or lymph nodes from mice were appropriately stained
by CD4-FITC, CD44-FITC, TCRβ-PE,
CD45-APC/Cy7, CD4-PE/Cy7, CD62L-biotin, and CD229.1-biotin in combination with streptavidin-APC. Generation of bone marrow chimera mice
CD3-depleted BM cells from Dsg3H1 mice were prepared by using CD3-microbeads (Miltenyi Biotech) and intravenously transferred into 7-Gy irradiated
129/SV mice or 129/SV Dsg3-/- mice. Two months
later, the recipient mice were used for further
experiments. Adoptive transfer
Dsg3-/- B cells were prepared from Dsg3-/- mice
as previously described (8). CD4+Vβ6+ T cells were
prepared from the spleen and LN of Dsg3H1 or Dsg3L3
mice by depleting B220+ and CD8+cells, followed by
the positive selection of Vβ6+ cells using the MACS
cell separation system (Miltenyi Biotech). 5× 106
Dsg3-/-B cells and 2.5 × 106 CD4+Vβ6+T cells were
intravenously transferred into Rag2-/- mice. In some
experiments, retrovirally transduced CD4+T cells were
transferred into Rag2-/- mice in combination with or
without Dsg3-/- B cells. Na¨ıve Ly9.1−CD4+ T cells
derived from wild type or Dsg3-/- mice with bone
marrow transfer from Dsg3H1 mouse were prepared
by depleting Ly9.1+, B220+, CD8+, Gr-1+, CD11b+,
DX5+, and CD44+ cells from splenocytes and LN
cells by magnetic beads and 3∼ 15 × 105T cells were
transferred with Dsg3-/-B cells into Rag2-/-mice.
Anti-Dsg3 antibody detection
Anti-Dsg3 IgG antiobody was quantified by ELISA and detected by living cell staining as described previously (7).
Histological analysis
Formalin-fixed tissue was stained with hema-toxylin and eosin, and observed with an inverted mi-croscope TE2000-U (Nikon, Tokyo, Japan). For
im-munofluorescent staining, 10-μm cryosections of the
palate were fixed with acetone, and subsequently
stained with anti-mouse IgG Ab-Alexa488 (Molecular
Probes, Eugene, OR), anti-TCRβ Ab-PE, and TOTO3
(Molecular Probes). Sections were observed under a confocal laser fluorescence microscope FV1000 (Olympus, Tokyo, Japan).
Results
Dsg3H T cell hybridoma cells respond to Dsg3 peptide
Nucleotide sequence of TCRα and β chain genes
derived from pathogenic Dsg3-reactive T cells were
analyzed and the results showed thatα chain consists
of AV8S13 and J21 and β chain consists of BV6S1
and Jβ1.3. T cell hybridoma cells (Dsg3H1 hybridoma
cell), in which these genes are reconstituted, were co-cultured with Dsg3 overlapping peptide and Dsg3H1 hybridoma cells respond to Dsg3 (301-315) peptide by IL-2 production (data not shown). Antigenic epitope of Dsg3H1 TCR was confirmed at peptide levels.
CD4+ T cells from Dsg3-specific TCR transgenic mouse respond to Dsg3 peptide
Using genes encoding specific TCR, Dsg3-spcific TCR transgenic mouse (Dsg3H1 mouse) was
generated. It was confirmed that Vβ6+ thymocytes
were able to develop to CD4+CD8− single positive
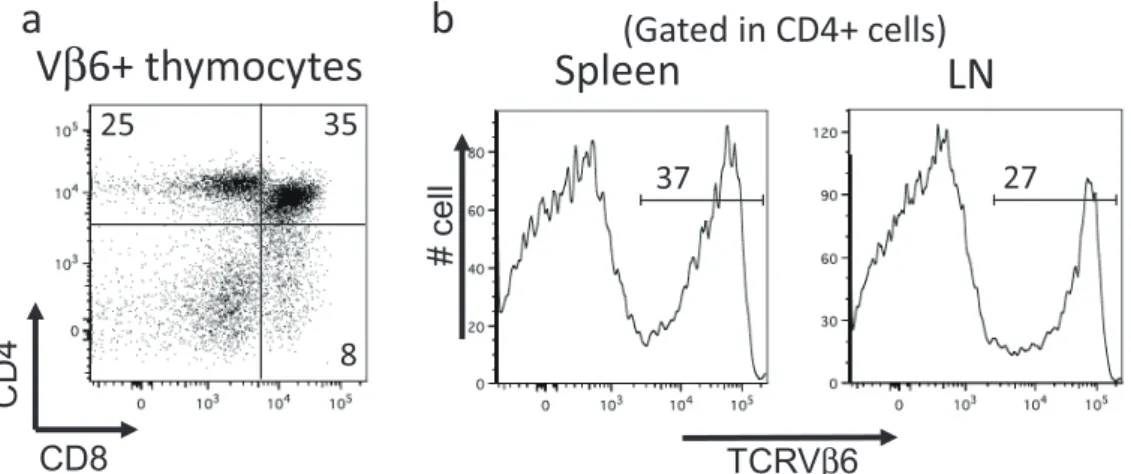
cells in the thymus (Figure 1a). In the secondary
lymphoid tissues, about 30% of CD4+cells were Vβ6+
cells, that is a higher proportion than that in wild type mouse (usually seen in less than 10%, Figure 1b). To evaluate whether these transgenic T cells are anergic or reactive to Dsg3, the splenocytes were cultured with antigenic Dsg3 peptide and Dsg3-specific response could be detected (Figure 2). Therefore, it was confirmed that Dsg3H1 mouse was expectedly generated harboring Dsg3-reactive T cells.
Transgenic T cells from Dsg3H1 mouse induce interface dermatitis in vivo
Since Dsg3H TCR was originally cloned from pathogenic Dsg3-reactive T cells that promoted anti-Dsg3 IgG production from B cells and pemphigus
CD4
V
β6+ thymocytes
CD8 25 35 8Spleen
LN
TCRVβ6 # cell(Gated in CD4+ cells)
37 27a
b
Figure 1. Flow cytometric analysis of thymus, spleen and lymph nodes (LN) in Dsg3H1 mouse. Single cell suspension was prepared from thymus (a), spleen and LN (b) of Dsg3H1 mouse and analyzed by flow cytometry. For the thymocytes analysis, dot plots analyzed by CD and CD8 expression were shown after gating on Vβ6+cells. For the analysis of spleen and LN, histograms for Vβ6 expression were shown after gating on CD4+cells. Dsg3H1 T cells developed into CD4+ single positive cells in thymus and detected as CD4+Vβ6+cells in the secondary lymphoid tissues. Data from reference (12).
0 1000 2000 3000 4000
1 2 Dsg3(301-315) Dsg3(223-237)
Splenocytes from Dsg3H1 mice
3H-thymidine uptake (cpm)
Figure 2. Dsg3-reactivity of splenocytes from Dsg3H1 mouse. Splenocytes were prepared from Dsg3H1 mouse and cultured with antigenic Dsg3 (301-315) peptide and control Dsg3 (223-237) peptide. cells were harvested 16 hours after3H-thymidine were added into culture medium and3H-thymidine uptake was evaluated. Dsg3H1 splenocytes responded to antigenic Dsg3 peptide. Data from reference (12).
phenotype in vivo after adoptive transfer with Dsg3
-/-B cells into Rag2-/-mouse, transgenic T cells (Dsg3H1
T cells) were expected to have the same pathogenicity. To confirm the expectation, Dsg3H1 T cells were isolated from Dsg3H1 mouse and transferred with
B cells into Rag2-/- mouse. The transferred mouse
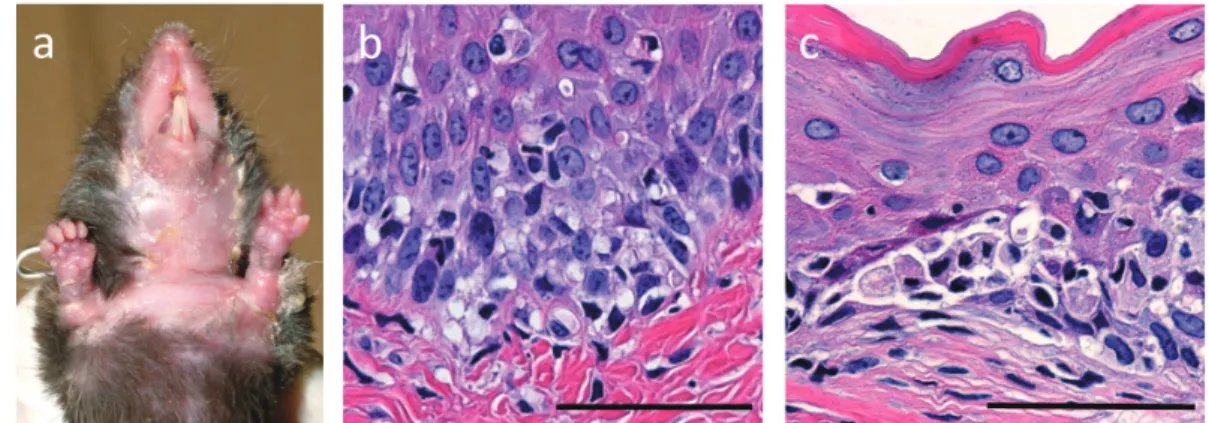
clinically developed erythema, hair loss and erosion in the skin as well as swelling in the limbs (Figure 3a) that started to appear around 2 - 3 weeks after the transfer. Histologically, acantholysis, a specific finding for pemphigus, was not observed in the palate and skin, instead many lymphocytes infiltrating at dermal-epidermal junction with liquefaction were observed (Figure 3b). In addition, lymphocytes further showed
intraepidermal infiltration and degeneration change of keratinocytes were observed (Figure 3c). These histological findings together demonstrated that the pathological change induced by Dsg3H1 T cells is interface dermatitis, a distinctively identified type of skin inflammation with unknown etiology. No anti-Dsg3 antibody production was detected by ELISA in the mouse (data not shown).
Development of Dsg3H1 T cells is regulated by tolerance mechanism against Dsg3
To analyze how Dsg3H1 T cells are influenced from tolerance mechanism against Dsg3, we evaluate
Figure 3. Pathogenic activity of Dsg3H1 T cells to induce interface dermatitis. CD4+T cells were prepared from Dsg3H1 mouse and transferred with Dsg3-/-B cells into Rag2-/-mouse. Skin phenotype were observed (a). Hair loss and erosion and swelling in the skin were apparently seen. Histopathology of the palate revealed that lymphocytes infiltrated at dermal-epidermal junction with liquefaction degeneration (b) and degenerated keratinocytes were observed (c). Scale bar; 50μm. Data from reference (12).
17%
97%
TCRV
β6
Dsg3 (+)
Dsg3 (-)
Figure 4. Dsg3H1 T cells undergo tolerance mechanism against Dsg3 and partial deletion. Bone marrow transfer was performed from Dsg3H1 mouse to wild type (left) or Dsg3-/-mouse (right). Thymocytes from recipient mice were analyzed by flow cytometry and histogram was shown after gating on CD4+cells. Almost all CD4+cells were Vβ6+cells in the absence of Dsg3. Only 20% of CD4+cells were detected as Vβ6+cells in the presence of Dsg3. The results clearly indicated that tolerance mechanism against Dsg3 exists in mouse. Data from reference (12).
the development of Dsg3H1 T cells in the presence or absence of Dsg3 after bone marrow cells were transferred from Dsg3H1 mouse into wild type or
Dsg3-/- mice. As a result, Dsg3H1 T cells fully
developed in the absence of Dsg3 since almost all
of CD4+ T cells were Vβ6+ cells in the lymph
nodes (Figure 4). On the other hand, Dsg3H1 T cells underwent partial deletion in the presence of
Dsg3 since only about 20% of CD4+ T cells were
Vβ6+ cells. Dsg3H1 mouse were further crossbred
with Rag2-/- mouse to avoid from endogenous TCR
expression and its development was similarly analyzed
by BMT into wild type or Dsg3-/-mice. Dsg3H1 T cells
could again fully develop in the absence of Dsg3, while the cells failed to develop and completely disappeared in the presence of Dsg3 (data not shown). These results demonstrated that tolerance mechanism against Dsg3 exists in mouse and Dsg3H1 mouse is a useful tool to clearly visualize the mechanism in future studies.
Discussion
The goal of this study is to clarify peripheral tolerance mechanism against Dsg3, an autoantigen of pemphigus, and provide the immunological basis for future development of antigen-specific immunotherapy. We generated Dsg3-specific TCR transgenic mouse, Dsg3H1 mouse, those transgenic T cells were able to undergo tolerance mechanism (12). Especially in the
setting of Rag2-/- background, Dsg3H1 T cells can
no longer develop in the presence of Dsg3 but fully develop in the absence of Dsg3, clearly showing “all or none” response. Using this mouse as sensor, it will be possible to clearly visualize where tolerance mechanism is functional, how much the mechanism can be manipulated and what kind of cells or molecules mediate the strict mechanism, by evaluating Dsg3H1 T cell development.
Through this study, we apparently demonstrated that Dsg3H1 mouse is a very useful mouse to monitor tolerance mechanism against Dsg3 and will utilize the mouse in further analysis for peripheral tolerance. Hopefully the new molecular or cellular pathway mediating peripheral tolerance would be discovered through this study and applied to creation of new idea for treatment of autoimmune diseases.
Conclusion
Dsg3-specific TCR transgenic mouse is useful to monitor tolerance mechanism against Dsg3.
Acknowledgements
This study was supported by the Waksman Foundation of Japan Inc.
References
(1) Amagai M, Klaus-Kovtun V and Stanley JR (1991) Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67: 869-877.
(2) Amagai M, Tsunoda K, Zillikens D, Nagai T
and Nishikawa T (1999) The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol 40: 167-170.
(3) Payne AS, Ishii K, Kacir S, Lin C, Li H, et al. (2005) Genetic and functional characterization of human pemphigus vulgaris monoclonal autoanti-bodies isolated by phage display. J Clin Invest 115: 888-899.
(4) Niizeki H, Inoko H, Mizuki N, Inamoto N, Watababe K, et al. (1994) HLA-DQA1, -DQB1 and -DRB1 genotyping in Japanese pemphigus vulgaris patients by the PCR-RFLP method. Tissue Antigens 44: 248-251.
(5) Ahmed AR, Mohimen A, Yunis EJ, Mirza NM, Kumar V, et al. (1993) Linkage of pemphigus vulgaris antibody to the major histocompatibility complex in healthy relatives of patients. J Exp Med 177: 419-424.
(6) Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, et al. (1991) Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proceedings of the National Academy of Sciences of the United States of America 88: 5056-5060. (7) Amagai M, Tsunoda K, Suzuki H, Nishifuji K,
Koyasu S, et al. (2000) Use of autoantigen-knockout mice in developing an active autoim-mune disease model for pemphigus. J Clin Invest 105: 625-631.
(8) Takahashi H, Amagai M, Nishikawa T, Fujii Y, Kawakami Y, et al. (2008) Novel system evaluating in vivo pathogenicity of desmoglein 3-reactive T cell clones using murine pemphigus vulgaris. J Immunol 181: 1526-1535.
(9) Hata T, Nishifuji K, Shimoda K, Sasaki T, Yamada T, et al. (2011) Transgenic rescue of desmoglein 3 null mice with desmoglein 1 to develop a syngeneic mouse model for pemphigus vulgaris. Journal of dermatological science 63: 33-39. (10) Kouskoff V, Signorelli K, Benoist C and Mathis D
(1995) Cassette vectors directing expression of T cell receptor genes in transgenic mice. J Immunol Methods 180: 273-280.
(1995) T cell proliferative response induced by DNA topoisomerase I in patients with systemic sclerosis and healthy donors. J Clin Invest 96: 586-596.
(12) Takahashi H, Kouno M, Nagao K, Wada N, Hata
T, et al. (2011) Desmoglein 3-specific CD4+ T
cells induce pemphigus vulgaris and interface dermatitis in mice. J Clin Invest 121: 3677-3688.
Helicobacter pylori regulate host non-coding RNA
expression to increase the proliferation of gastric
epithelium during chronic infection
Hitomi Mimuro
Division of Bacteriology, Department of Infectious Diseases Control, International Research Center for Infectious Diseases, Institute of Medical Science, The University of Tokyo 4-6-1, Shirokanedai, Minato-ku, Tokyo 108-8639, Japan
Introduction
Chronic inflammation is a critical risk factor for and has been estimated to cause 20% of all human cancers. Specific pathogens have been identified as causal risks for particular types of cancer; Helicobacter pylori in gastric cancer, Hepatitis B and C viruses in liver cancer, Papilloma virus for ovarian cancer, EB virus for malignant lymphoma, adult T-cell leukemia virus for ATL, and human herpes virus 8 for Ka-posi’s sarcoma. However, the molecular mechanisms by which these microbial infections trigger malig-nancies remain largely unknown. Understanding these mechanisms would therefore contribute to the molecu-lar basis for development of novel preventive, therapeu-tic, and diagnostic approaches against cancer.
Chronic Helicobacter pylori (Hp) infection of the gastric epithelium is strongly associated with the development of gastritis, peptic ulcers, mucosa-associated lymphoid tissue lymphoma, and gastric cancer. In gastric cancer, nearly 60% of all cases in developed countries and 75% in developing countries are attributed to chronic infection with Hp. During persistent Hp colonization of the gastric mucosa, sustained inflammation and aberrant epithelial proliferation are considered to be major factors that lead to Hp-associated gastric diseases, although the mechanisms underlying disease progression remain elusive.
An accumulation of genetic and epigenetic alterations in normal tissues triggers carcinogenesis. For example, DNA methylation of specific genes can be found in up to several percent of the cells in noncancerous gastric mucosae, and the rate of gene methylation is considered to be correlated with an increased risk of gastric cancer. Importantly, Hp infection in the stomach has been shown to potentiate the induction of aberrant DNA methylation in the gastric epithelium, along with chronic inflammation. Notably, methylation of promoter sites leads to gene expression silencing.
On the other hand, cumulative evidence suggests that microRNAs (miRNAs) might play important roles in the initiation and progression of various
human diseases. In this context, we sought to
identify miRNAs that are causally involved in gastric malignancies associated with Hp infection by using several systematic and bioinformatic approaches and studying gastric epithelial cell lines, stomach tissue from Mongolian gerbils, and human stomach biopsy specimens. In this comprehensive study, we demonstrate that miR-210 is a critical miRNA, which regulates gastric epithelial cell proliferation by targeting potential oncogenes. Furthermore, our work provides substantial evidence for the causal involvement of epigenetic silencing of miR-210 in development of gastric malignancies induced by chronic Hp infection.