Studies on the Electronic Structures of Model Compounds of p-Doped Polythiophene
Masaki Tateno
A DISSERTATION
Submitted in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY (SCIENCE)
Tokyo Metropolitan University Tokyo, Japan
2013
Contents.
Chapter 1. General Introduction.
1-1. Conductive Polymer ... 1
1-2. Previous Studies on p-doped Polythiophene using Oligomers as a Model ... 2
1-3. Functional -Conjugated Oligomers ... 3
1-4. Out Line ... 4
1-5. References ... 6
Chapter 2. Steric Control in the π-Dimerization of Oligothiophene Radical Cations Annelated with Bicyclo[2.2.2]octene Units. Abstract ... 9
2-1. Introduction ... 10
2-2. Molecular Design ... 12
2-3. Calculation Methods ... 13
2-4. Synthesis ... 14
2-5. Results ... 16
2-6. Discussion ... 27
2-7. Conclusion ... 34
2-8. Experimental Section ... 35
2-9. References ... 41
Chapter 3. The Conductivities of Gold-Nanoparticles Protected by Bicyclo[2.2.2]octene Annelated Oligothiophenes. Abstract ... 45
3-1. Introduction ... 46
3-2. Molecular Design ... 48
3-3. Synthesis ... 49
3-4. Properties of Oligothiophenes Capped with Methylthio Groups ... 51
3-5. Properties of AuNPs Covered with Oligothiophene ... 57
3-6. Discussion ... 60
3-7. Conclusion ... 62
3-8. Experimental Section ... 62
3-9. References ... 71
Chapter 4. Biradical Character of Linear -Conjugated Oligomer Dications Composed of Thiophene, Pyrrole, and Methylthio End-Capping Units. Abstract ... 74
4-1. Introduction ... 75
4-2. Evaluation Methods of singlet biradical character ... 75
4-3. Molecular Design ... 76
4-4. DFT Calculations ... 77
4-5. Synthesis ... 83
4-6. Results ... 84
4-7. Conclusion ... 92
4-8. Experimental Section ... 92
4-9. References ... 96
List of Publications ... 99
Acknowledgement ... 100
- 1 -
Chapter 1. General Introduction.
1-1. Conductive Polymer.
Conductive polymers are the polymers having electronic conductance. It had been thought that electronic conductance was the specific properties of metals which have free electrons, while polymers, which are organic materials having no free electron, were insulators. However, in 1970’s, Shirakawa and co-workers reported the electronic conductance of iodine-doped polyacetylene (Figure 1-1),1 and then, the conductive polymers have been extensively studied.
Figure 1-1. The structure of polyacetylene.
Although doped-polyacetylene shows high conductivity, which is comparable to that of metals (102 S/cm), polyacetylene has some disadvantages that are instable, insoluble and hard-to-melt for the practical applications. On the other hand, -conjugated polymers containing aromatic rings (Scheme 1-1), which include poly-p-phenylene,2 polythiophene,3 polypyrrole,4 polyaniline5 and polyphenylenevinylene,6 have high stability and the possibility to become highly soluble by the introducing substituent groups. Thus, these -conjugated polymers have been synthesized and studied extensively.
Scheme 1-1. Schematic illustrations of -conjugated polymers containing aromatic rings.
Polythiophene, one of these -conjugated polymers, and its derivatives have been studied extensively due to its high stability and conductivity. It is known that polythiophenes in highly p-doped state show high conductivity and no ESR signal.7 It had been thought that the possible
- 2 -
charge careers of p-doped polythiophenes were only polaron and bipolaron, which were radical cation and closed-shell dication in chemical term, respectively (Figure 1-2), and bipolaron was taken as the charge career of polythiophenes in highly p-doped states due to its ESR-silent character. Thus, cationic species of -linked linear oligothiophenes have been intensively investigated as models of p-doped states of conducting polythiophenes.
(a)
(b)
Figure 1-2. The illustrations of (a) polaron and (b) bipolaron.
1-2. Previous Studies on p-doped Polythiophene using Oligomers as a Model.
Experimental and theoretical studies on -conjugated oligomers have also been conducted for the purpose of revealing detailed conduction mechanism of conductive polymers.
In 1992, Miller and co-workers first reported -dimer of oligothiophene radical cations by the using of terthiophene, which has methyl group at the both -positions to stabilize its oxidized states (Scheme 1-2).8 It was proposed that -dimers, formed by intermolecular dimerization of two polarons with ESR inactive properties, are alternative to bipolarons. Experimental studies conducted thus far on the π-dimers of oligothiophene radical cations have revealed chain length dependence9 and solvent effects10 on π-dimerization in the solution phases and the structures of the π-dimer in crystalline form.11 Furthermore, detailed studies on the dications of bridged oligothiophene dimers12 and thienoacene radical cations13 as models of π-dimers have also been reported.
Scheme 1-2. Schematic illustration of -dimerization of dimethylterthiophene radical cations.
On the other hand, Janssen and co-workers proposed that charge careers of p-doped polythiophenes were “polaron pair”, which have two polarons in one molecule (called as singlet
- 3 -
biradical in chemical term). Their proposal was based on the evidence that oxidized states of duodecithiophene (Figure 1-3) did not show any signals by the ESR measurements using various equivalence of oxidants.14 Whereas, recent theoretical studies demonstrated that oligothiophene dications composed of four (TDB3P86-30%)15 or six (B3LYP)16 thiophene rings have a singlet biradical character. However, it was pointed out that the absorption spectrum of the sterically unhindered duodecithiophene dication showing two broad absorption bands, which were previously assigned as intrachain polaron pair,14 is totally different from that of a sterically hindered duodecithiophene dication which shows only one broad absorption band.17 The discrepancy between sterically unhindered and hindered oligomers may be brought about by the presence and absence of the intermolecular - interaction but the cause remains unclear. This ambiguity stems partly from lack of information about solid state properties of the dications.
Figure 1-3. The structures of oligothiophenes in ref 14.
1-3. Functional -Conjugated Oligomers.
Although studies on -conjugated oligomers had been mainly conducted as model systems of polymers, it was shown in recent decades that -conjugated oligomers show good photo- and electro-properties due to their regular-structures unlike polymers. Because of these properties of
-conjugated oligomers, applications of -conjugated oligomers for field effect transistor (FET), organic light emitting diodes (OLED) and dye-sensitized solar cells (DSSC) have been extensively studied (Figure 1-4).18-20 Also in our laboratory, several functional -conjugated oligomers have been reported.21
The organic-inorganic composite materials using these -conjugated molecules have also been extensively studied, for example, metal-nanoparticles (MNPs)22,23 and self-assembled monolayers (SAMs).24 Most of these studies aim the functional materials using conductivity of
-conjugated molecules between metals, and the gold-nanoparticles (AuNPs) covered with oligothiophenes have attracted much attention (Figure 1-5).
- 4 - (a)
(b)
(c)
Figure 1-4. The examples of -conjugated oligomers for (a) FET,18 (b) OLED19 and (c) DSSC.20
(a) (b)
(c)
Figure 1-5. The examples of -conjugated molecules using as spacer units between AuNPs.23
Because of these background described above, revealing the electronic structures of
-conjugated oligomers is important for application of -conjugated oligomers more than just understanding of conduction mechanism of polymers.
1-4. Outline.
In this work, the model compounds of -dimers and singlet biradicals, which are regarded as charge careers of p-doped polythiophenes, were synthesized, and their electronic structures of oxidized states were studied. The AuNPs covered with oligothiophene having bicyclo[2.2.2]octene (BCO) group(s) were also investigated for the purpose of developing conduction-switching devices.
- 5 -
The oligothiophenes 2-6 (Figure 1-6) having BCO groups, which stabilize oxidized states of oligothiophenes, were designed and synthesized as the models of -dimers, and their radical cation salts were isolated. Oligothiophenes 2 and 3 having one and two extra thiophene ring(s) more than previously-reported terthiophene 125 were designed for the aim of investigating the effect of chain length on -dimerization. In comparison among oligothiophenes 2 and 4-6 being constructed with the same number of thiophene rings but having various number of BCO groups at various positions, it is expected that the area of -systems to interact in -dimerizations and the structures of
-dimers were different from each other. Thus, these oligothiophenes 1-6 would provide many evidences of -dimerization of oligothiophene radical cations.
Figure 1-6. The structures of 1-6.
The BCO-annelated oligothiophenes 7 and 8 (Figure 1-7) were designed, synthesized and introduced to AuNPs for the purpose of developing conduction-switching devices. The terthiophenes 9 were also synthesized and introduced to AuNPs for the comparison with 7 and 8. For the AuNPs covered with BCO-annelated oligothiophenes, it is expected that their conductions in neutral state are suppressed by elongating the distances between AuNPs due to steric repulsions between BCO groups, while their conductions in p-doped state are promoted by the interactions like
-dimerizations between unsubstituted thiophene rings.
Figure 1-7. The structures of 7-9.
The -conjugated oligomers 10-13 were designed based on the thiophene-pyrrole mixed-oligomers DHnTP, which were previously reported its FET properties in our group,21a as the
- 6 -
model of singlet biradical (Figure 1-8). From the calculations, the biradical index (see Chapter 4) of the dications of oligomers 10-13 showed larger values than the corresponding dications of oligothiophenes, respectively. The oligomers 10-13 have two long alkyl chain at the nitrogen of pyrroles for the enhancement of solubility and two methylthio groups at the both ends for the stabilizing their oxidized states.
a)
b)
Figure 1-8. The structures of (a) DHnTP and (b) 10-13.
1-5. References.
(1) H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang, A. J. Heeger, J. Chem. Soc., Chem.
Commun.1977, 578.
(2) (a) L. M. Goldenberg, P. C. Lacaze, Synth. Met. 1993, 58, 271. (b) C. G. Andrew, K. Müllen, Adv. Polym. Sci. 2006, 199, 1.
(3) (a)J. Roncali, Chem. Rev. 1992, 92, 711. (b) M. Leclerc, K. Faid, Adv. Mater. 1997, 9, 1087.
(4) (a) B. R. Saunders, R. J. Fleming, K. S. Murray, Chem. Mater. 1995, 7, 1082. (b) Y.
Berdichevsky, Y.-H. Lo, Adv. Mater. 2006, 18, 122. (c) A. Ramanavičius, A. Ramanavičienė, A.
Malinauskas, Electrochim. Acta. 2006, 51, 6025.
(5) (a) E. M. Geniès, A. Boyle, M. Lapkowski, C. Tsintavis, Synth. Met. 1990, 36, 139. (b) J.
Huang, S. Virji, B. H. Weiller, R. B. Kaner, Chem. Eur. J. 2004, 10, 1314. (c) N.-R. Chiou, A. J.
Epstein, Adv. Mater. 2005, 17, 1679.
(6) (a) H. C. F. Martens, P. W. M. Blom, H. F. M. Schoo, Phys. Rev. B 2000, 61, 7489. (b) I. N.
Hulea, H. B. Brom, A. J. Houtepen, D. Vanmaekelbergh, J. J. Kelly, E. A. Meulenkamp, Phys.
Rev. Lett. 2004, 93, 166601. (c) A. Kumar, P. K. Bhatnagar, P. C. Mathur, M. Husain, S.
Sengupta, J. Kumar, J. Appl. Phys. 2005, 98, 024502.
(7) (a) J. L. Brédas, G. B. Street, Acc. Chem. Res. 1985. 18, 309. (b) K. Kaneto, S. Hayashi, S. Ura, K. Yoshino, J. Phys. Soc. Jpn. 1985, 54, 1146. (c) J. Chen, A. J. Heeger, F. Wudl, Solid State Commun. 1986, 58, 251.
- 7 -
(8) (a) M. G. Hill, K. R. Mann, L. L. Miller, J. –F. Penneau, J. Am. Chem. Soc. 1992, 114, 2728. (b) D. D. Graf, J.P.Campbell, L.L.Miller, K.R.Mann, J. Am. Chem. Soc. 1996, 118, 5480.
(9) (a) P. Bäuerle, U. Segelbacher, A. Maier, M. Mehring, J. Am. Chem. Soc. 1993, 115, 10217-10223; (b) P. Bäuerle, U. Segelbacher, K.-U. Gaudl, D. Huttenlocher, M. Mehring, Angew. Chem. 1993, 105, 125–127; Angew. Chem., Int. Ed. Engl. 1993, 32, 76-78.
(10) Y. Yu, E. Gunic, B. Zinger, L. L. Miller, J. Am. Chem. Soc. 1996, 118, 1013-1018.
(11) (a) D. D. Graf, J. P. Campbell, L. L. Miller, K. R. Mann, J. Am. Chem. Soc. 1996, 118, 5480-5481; (b) D. D. Graf, R. G. Daun, J. P. Campbell, L. L. Miller, K. R. Mann, J. Am. Chem.
Soc. 1997, 119, 5888-5899.
(12) (a) T. Kaikawa, K. Takimiya, Y. Aso, T. Otsubo, Org. Lett. 2000, 2, 4197-4199; (b) T. Satou, T.
Sakai, T. Kaikawa, K. Takimiya, T. Otsubo, Y. Aso, Org. Lett. 2004, 6, 997-1000; (c) T. Sakai, T.
Satou, T. Kaikawa, K. Takimiya, T. Otsubo, Y. Aso, J. Am. Chem. Soc. 2005, 127, 8082-8089;
(d) J. Casado, K. Takimiya, T. Otsubo, F. J. Ramírez, J. J. Quirante, R. P. Ortiz, S. R. González, M. M. Oliva, J. T. López Navarrete, J. Am. Chem. Soc. 2008, 130, 14028-14029; (e) K. M.
Knoblock, C. J. Silvestri, D. M. Collard, J. Am. Chem. Soc. 2006, 128, 13680-13681.
(13) (a) R. M. Osuna, M. C. R. Delgado, V. Hernández, J. T. López Navarrete, B. Vercelli, G. Zotti, J.
J. Novoa, Y. Suzuki, S. Yamaguchi, J. T. Henssler, A. J. Matzger, Chem. Eur. J. 2009, 15, 12346-12361; (b) C. C. Ferrón, M. C. R. Delgado, V. Hernández, J. T. López Navarrete, B.
Vercelli, G. Zotti, M. C. Cortada, J. J. Novoa, W. Niu, M. He, F. Hartl, Chem. Commun. 2011, 47, 12622-12624.
(14) J. A. E. H. van Haare, E. E. Havinga, J. L. van Dongen, R. A. J. Janssen, J. Cornil, J-L. Brédas, Chem. Eur. J. 1998, 4, 1509.
(15) U. Salzner, J. Phys. Chem. A 2008, 112, 5458.
(16) (a) S. S. Zade, M. Bendikov, J. Phys. Chem. B 2006, 110, 15839. (b) Y. Gao, C.-G. Liu, Y.-S.
Jiang, J. Phys. Chem. A 2002, 106, 5380.
(17) (a) Y. Ie, A. Han, T. Otsubo, Y. Aso, Chem. Commun. 2009, 3020. (b) T. Kurata, T. Mohri, K.
Takimiya, T. Otsubo, Bull. Chem. Soc. Jpn. 2007, 80, 1799.
(18) M. Halik, H. Klauk, U. Zschieschang, G. Schmid, S. Ponomarenko, S. Kirchmeyer, W. Weber, Adv. Mater. 2003, 15, 917.
(19) T. Noda, H. Ogawa, N. Noma, Y. Shirota, J. Mater. Chem. 1999, 9, 2177.
(20) N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki, K. Hara, J. Am. Chem. Soc. 2006, 128, 14256.
(21) (a) M. Fujii, T. Nishinaga, M. Iyoda, Tetrahedron Lett. 2009, 50, 555. (b) M. W-Harry, A.
Bhaskar, G. Ramakrishna, T. Goodson, M. Imamura, A. Mawatari, K. Nakao, H. Enozawa, T.
Nishinaga, M. Iyoda, J. Am. Chem. Soc. 2008, 130, 3252. (c)T. Nishinaga, T. Miyata, M. Tateno, M. Koizumi, M. Takase, M. Iyoda, N. Kobayashi, Y. Kunugi, J. Mater. Chem. 2011, 21, 14959.
- 8 -
(22) (a) M.-C. Daniel, D. Astruc, Chem. Rev. 2004, 104, 293. (b) A. Zabet-Khosousi, A.-A. Dhirani, Chem. Rev. 2008, 108, 4072.
(23) (a) S. Taniguchi, M. Minamoto, M. M. Matsushita, T. Sugawara, Y. Kawada, D. Bethell, J.
Mater. Chem. 2006, 16, 3459. (b) G. Zotti, B. Vercelli, A. Berlin, Chem. Mater. 2008, 20, 397.
(24) J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo, G. M. Whitesides, Chem. Rev. 2005, 105, 1103.
(25) D. Yamazaki, T. Nishinaga, N. Tanino, K. Komatsu, J. Am. Chem. Soc. 2006, 128, 14470.
- 9 -
Chapter 2.
Steric Control in the π-Dimerization of Oligothiophene Radical Cations Annelated with Bicyclo[2.2.2]octene Units.
Abstract
Series of oligothiophenes 2(nT) (n = 4,5) annelated with bicyclo[2.2.2]octene (BCO) units at both ends and quaterthiophenes 3a–c annelated with various numbers of BCO units at different positions were newly synthesized to investigate the driving forces of -dimerization and the structure–property relationships of the -dimers of oligothiophene radical cations. Their radical cation salts were prepared via a chemical one-electron oxidation using nitrosonium hexafluoroantimonate. From variable temperature electron spin resonance and electronic absorption measurements, the -dimerization capability was found to vary among the members of the 2(nT)+•SbF6–
series and 3+•SbF6–
series of compounds. To examine these results, density functional theory (DFT) calculations at the M06-2X/6-31G(d) level were conducted for the -dimers. This level of theory was found to successfully reproduce the previously reported X-ray structure of (2(3T))22+
having a bent -dimer structure with cis-cis conformations. The absorption bands obtained by time-dependent DFT calculations for the -dimers were in reasonable agreement with the experimental spectra. The attractive and repulsive forces for the π-dimerization were divided into four factors: (1) SOMO–SOMO interactions, (2) van der Waals forces, (3) solvation, and (4) Coulomb repulsion, and the effects of each factor on the structural differences and chain length dependence are discussed in detail.
- 10 -
2-1. Introduction.
In recent years, the properties of -dimers formed by dimerizations of radical species have attracted much attentions again. The formation and dissociation of -dimers are reversible in concentrated and/or low-temperature solution phases, and the -dimers are known to be singlet species due to their ESR-inactive characters. Phenalenyl radicals, tetrathiafulvalene (TTF) cation radicals, tetracyanoethylene (TCNE) anion radicals, tetracyanoquinodimethane (TCNQ) anion radicals and viologen cation radicals are well known to form -dimers and have been extensively studied.1-5
The -dimer of oligothiophene radical cation was first reported by Miller and co-workers in 1992,6 and has attracted much attention because -dimers would be alternative to bipolarons as described in Chapter 1. Bäuerle and co-workers reported the -dimerizations of radical cations of ECnT, which are oligothiophenes having cyclohexyl groups at the both ends, and DD6T, which is sexithiophene having two dodecyl groups, in dichloromethane solution (Figure 2-1). From the comparison among ECnT, DD6T and DM3T, it was shown that their -dimerization abilities of the oligothiophene radical cations increased with increasing chain length (chain length dependence).7 In the study using oligothiophenes having methoxy groups MeOnT (Figure 2-2(a)), it was reported that
-dimerizations were promoted in acetonitrile relative to those in dichloromethane solution.8 First X-ray crystal structure of -dimer of oligothiophene radical cation was reported by the using of terthiophene capped with phenyl groups at the both ends to stabilize the oxidized states (Figure 2-2(b)).9
(a) (b)
Figure 2-1. The structures of (a) ECnT and (b) DD6T.7
(a) (b)
Figure 2-2. The structure of (a) MeOnT8 and (b) DP3T.9
- 11 -
On the other hand, detailed properties of -dimers of oligothiophene radical cations were also reported by the using of bridged-oligothiophenes that were easy to form -dimers. Aso and co-workers synthesized the quinquethiophenes bridged by alkyl groups at the both ends or one end, and reported properties of the dications of the oligothiophene.10 Collard and coworkers also reported the properties of the -dimers of the oligothiophenes bridged at the -positions of central thiophene rings (Figure 2-3).11 The -dimers of thienoacenes which are constructed with fused-thiophene rings was also reported.12 However, the dominant factors of -dimerizations have not been revealed in previous studies (Figure 2-4).
(a) (b)
Figure 2-3. The structures of bridged oligothiophenes (a) in ref.10 and (b) in ref 11.
Figure 2-4. The structures of thienoacenes.12
- 12 -
Many studies on -dimers of oligothiophene radical cations have been reported as mentioned above, while theoretical studies on the -dimers have been limited.13 One of the reasons for this is that the most popular density functional theory (DFT) method, B3LYP,14 is poor at estimating weak intermolecular interactions, and is not suitable for the study of most -dimers. Thus, to our knowledge, no detailed studies based on both experiments and theoretical calculations for the quantitative estimation of the attractive and repulsive intermolecular interactions in the π-dimers of oligothiophene radical cations in solution have been performed. On the other hand, in recent years, there has been growing interest in π-dimer-based supramolecular chemistry. Catenanes,2b rotaxanes,5b actuators15 and ball bearings16 using π-dimer formations as the driving force have been demonstrated. Therefore, the detailed quantitative estimation of the π-dimerization of oligothiophene radical cations would provide valuable guidance for the further development of π-dimer-based supramolecular chemistry.
2-2. Molecular Design.
Unsubstituted oligothiophene radical cations are reactive and unstable species, which are different from phenalenyl radicals and TTF cation radicals. Thus, oligothiophene radical cations were used as intermediates for the synthesis of polythiophenes.17 Thus, it is important to design oligothiophenes to stabilize their radical cations for detailed investigations of their -dimers. One of the general methods stabilizing the radical cations is to introduce the substituents stabilizing oxidized states, for example alkyl, phenyl and methylthio groups, at the most reactive terminal
-positions. However, the stabilities of the radical cations of DM3T6 and ECnT7a were not very high.
In contrast, the radical cation of DP3T having phenyl groups had very high stability that enabled X-ray crystallography,9 but the substituents that may alter the electronic structure of oligothiophene like phenyl and methylthio groups were undesirable. On the other hand, in our laboratory, the cationic species of oligothiophenes SnTBCO (Figure 2-5) stabilized with the annelation of bicyclo[2.2.2]octene (BCO) units were shown to be suitable for X-ray crystallography.18 The BCO units are an alkyl group while they highly stabilize the oxidized states by the combined effects of steric protection, hyperconjugation, and inductive electron donation.19 The BCO units suppress intermolecular interactions of -dimer due to its bulky frameworks. Actually, the radical cations of SnTBCO fully annelated with BCO units did not form -dimers in both solution and crystalline phases.18 In sharp contrast, the radical cation salt of terthiophene 1, which has BCO groups only at its ends, showed a bent π-dimer.20 These results suggest that π-dimerization could be controlled by the use of BCO groups. Thus, in the present study, quaterthiophene 2 and quinquethiophene 3, which have BCO groups at both ends, as well as quaterthiophenes 4–6, which have various numbers of BCO groups at various positions were designed and synthesized (Figure 2-6). In a series of 1-3, the
- 13 -
formation of bent π-dimers are expected; the structures are different from the previously reported π-dimers of DM3T,6 EC4T and EC5T,7a which lack bulky groups. In quaterthiophenes 4–6, each π-dimer would have different π-contact areas. Therefore, the effects of structure and π-contact areas on π-dimerization can be estimated from the comparison of these systems.
Figure 2-5. The structures of SnTBCO.
Figure 2-6. The structures of 1-6.
2-3. Calculation Methods.
As mentioned above, theoretical studies on the -dimers of oligothiophene radical cations have been limited.13 On the other hand, Miller and Novoa conducted theoretical studies on -dimers of phenalenyl radicals, TCNE anion radicals, and TCNQ anion radicals, and considered the interactions in -dimerization caused by the overlap of the SOMOs of the radical species to be
“multicenter two-electron bonds”.1d,3d,4,13b
However, the calculations they used in these studies were higher level of theories than B3LYP method, and it is difficult to apply these calculations to the larger molecules in this study. In the studies of -dimers of thienoacene radical cations, the calculations of -dimers using B3LYP levels were successfully conducted in the presence of counter-anions.12 However, it is predicted that the inclusion of counter-ions in the calculations make it difficult to find the global minimum structures of -dimers due to its high degrees of freedom.
Furthermore, the counter-anion using in this study, hexafluoroantimonate anion (SbF6
-), is weakly coordinating anion, and it has been reported that counter ions have little effect on the spectral properties of -dimeric species and the thermodynamic parameters for the -dimerization of 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) radical anion.21 Thus, in this study, the calculations, which reproduced the structures and thermodynamic parameters of -dimers well even
- 14 -
in the absence of counter-ions, were conducted. First, CAM-B3LYP22 and B3LYP-D23 methods that the modified B3LYP methods improved the estimation of the weak intermolecular interactions were tested using (1)2
2+. However, these methods gave calculatedHdim values (+15.0 kcal mol–1 and -19.5 kcal mol–1, respectively) that were significantly different from the experimental value (-6.7 kcal/mol21). Secondly, M06-2X24 methods, which can better estimate the energies of weak intermolecular interactions than the B3LYP method and has recently been described as efficient for the calculations of the -dimers of the radicals of 1,2,3,5-dithiadiazolyl and 1,2,3,5-diselenadiazolyl,25 were tested. As the results, the calculatedHdim value (-8.9 kcal/mol) for (1)2
2+ was in reasonable agreement with the experimental value and the optimized structure was found to successfully reproduce the previously reported X-ray structure of (1)2
2+ (Figure 2-7).20 Thus, in this study, M06-2X/6-31G(d) level of theory was used.
(a)
(b)
Figure 2-7. (a) Top and side views of optimized structures of (1)22+
calculated at the M06-2X/6-31G(d) level and (b) X-ray structure of (1)22+
.20
2-4. Synthesis.
The synthesis of 2 and 3 was performed using conventional cross-coupling reactions. As shown in Scheme 2-1, 2, 3 and 4 were synthesized by the Stille coupling of 2-(trimethylstannyl)- 4,5-bicyclo[2.2.2]octenothiophene (14)18a with the corresponding brominated oligothiophenes 15,26 1627 and 17.28 Bromoterthiophene 22 was synthesized by a bromination reaction between N-bromosuccinimide (NBS) and terthiophene 21, which was obtained by the Stille coupling of 2-(trimethylstannyl)-3,4-bicyclo[2.2.2]octenothiophene (19) with 5-bromo-5’-methyl-2,2’- bithiophene (20).29 Then, 5 was synthesized by the Stille coupling of 14 with 22. Bromoterthiophene 26 was also synthesized from terthiophene 25, which was obtained from 5-bromo-3,4:3’,4’-
2.95 Å
2.98 Å
- 15 -
bis(bicyclo[2.2.2]octeno)-2,2’-bithiophene (23)18a and 2-methyl-5-(trimethylstannyl)thiophene (24),30 and 6 was synthesized by the Stille coupling of 14 with 26.
Scheme 2-1. Synthesis of 2-6.
- 16 -
2-5. Results.
Optical and electronic properties in neutral states.
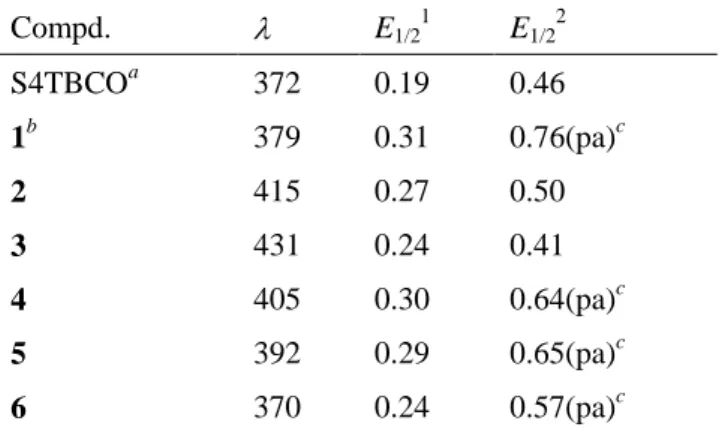
The electronic absorption spectra of 2-6 in dichloromethane are summarized in Figure 2-8 and Table 2-1, together with the data of S4TBCO.18a The longest wavelength absorptions of 1-3 were bathochromically shifted in accordance with the extension of the -systems. In contrast, in the comparison of the quaterthiophenes S4TBCO, 2, and 4-6, hypsochromic shift of the longest wavelength absorption bands and a decrease in the extinction coefficient with increasing numbers of BCO units at the 3,4-position of thiophene rings were observed. The distortion in the -systems caused by the steric repulsion between bridgehead protons and sulfur atoms is responsible for these phenomena, as mentioned in our previous report.18a
Figure 2-8. Absorption spectra of 2–6 in dichloromethane.
Table 2-1. Longest Wavelength Absorption Bands (/nm) and First and Second Oxidation Potentials (E/V vs Fc/Fc+) of S4TBCO and 1–6 in Dichloromethane.
Compd. E1/2
1 E1/2
2
S4TBCOa 372 0.19 0.46
1b 379 0.31 0.76(pa)c
2 415 0.27 0.50
3 431 0.24 0.41
4 405 0.30 0.64(pa)c
5 392 0.29 0.65(pa)c
6 370 0.24 0.57(pa)c
aRef.18a. bRef. 20. cAnodic peak potential in irreversible wave.
- 17 -
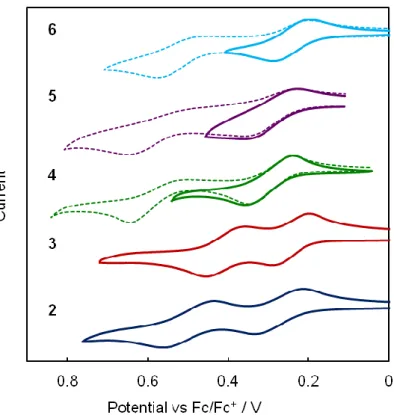
To examine the redox behavior of 2–6, cyclic voltammetry was conducted in dichloromethane with tetra-n-butylammonium perchlorate as the electrolyte. The voltammograms are shown in Figure 2-9 and the values of the oxidation potentials are summarized in Table 2-1, together with those of S4TBCO.18a All of the compounds exhibited reversible first oxidation waves, and 2 and 3, which have BCO units at both ends, exhibited reversible second oxidation waves. In contrast, 4–6, which have BCO units at one end and methyl groups at the other end, exhibited irreversible second waves. These results indicate that the annelation of BCO units at both ends is important for the stabilization of the dication of oligothiophenes. In 1-3, the oxidation potential decreased with the extension of the π-systems by increase in the thiophene rings, which was similar to the case of the absorption spectra described above. In the comparison of the quaterthiophenes S4TBCO, 2, and 4–6, an increasing number of annelations with electron-donating BCO units caused a decrease in the oxidation potential irrespective of the twisted structure in the neutral state, as mentioned above.
Figure 2-9. Cyclic voltammograms of 2–6 in dichloromethane.
Properties of Radical Cations.
On the basis of the reversibility of the first oxidation process observed using cyclic voltammetry, radical cations of 2–6 were expected to be stable. Thus, one-electron oxidation of 2–6
- 18 -
were conducted in dichloromethane using nitrosonium hexafluoroantimonate as the oxidant, and the radical cation salts (> 95% purity) was obtained as precipitates by adding hexane to the solution. All of the radical cation salts were stable and can be handled under ambient conditions. Unfortunately, however, the single crystals suitable for X-ray analysis have not been obtained so far.
ESR measurements of 2+•SbF6–
- 6+•SbF6–
were conducted in dichloromethane solution. As shown in Figure 2-10, all of the radical cation salts showed ESR signals at room temperature. For all of the radical cations except 6+•SbF6–
, the intensity of the signals decreased as temperature decreased, and the spectral changes were fully reversible. These results indicate that the doublet radical cations formed singlet species at low temperatures. In contrast, such spectral changes were not observed for the signal of 6+• under the measurement conditions (1.5 mM, 183 K).
(a) (b) (c)
(d) (e)
Figure 2-10. ESR spectra of (a) 2+•SbF6–
(1.0 mM), (b) 3+•SbF6–
(0.1 mM), (c) 4+•SbF6–
(0.1 mM), (d) 5+•SbF6–
(1.5 mM) and (e) 6+•SbF6–
(1.5 mM).
In 2+•, the simulation of ESR was also conducted by the using of calculated coupling constants at the B3LYP/6-31G(d) level. The Simulated ESR spectrum of 2+• (aH = 0.86 G (4H), 0.55 G (2H), 0.42 G (4H)) was good agreement with experimental ESR spectrum (Figure 2-11).
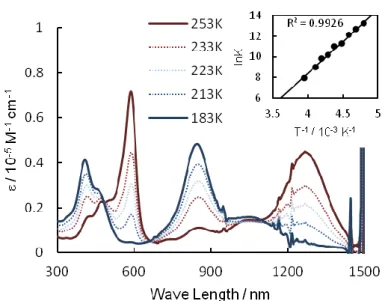
Next, the electronic absorption spectra of 2+•SbF6–
- 5+•SbF6–
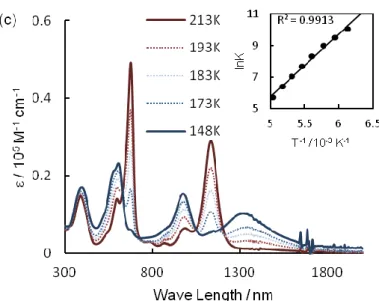
were measured at various temperatures. As shown in Figures 2-12- 2-15, all of the radical cation salts showed two absorption bands near room temperature; the lower and higher energy bands were assigned as the HOMO–SOMO and SOMO–LUMO transitions, respectively.31 The intensity of these two bands decreased with decreases in temperature, and three new absorption bands appeared. These spectral changes were also reversible and consistent with those previously observed in the π-dimer formation
- 19 -
of oligothiophene radical cations.6,7,10 Thus, it was confirmed that 2+•- 6+• formed -dimers at low temperatures. The dimerization equilibrium constants were estimated from these measurements using the absorption of the monomer in the higher energy region, considering that only the following equilibrium was present judging from the appearance of isosbestic points:
2 nT+• ⇄ (nT)2 2+
The dimerization enthalpy (Hdim) and entropy (Sdim) were then obtained by van’t Hoff plots. The results for the absorption bands of the monomer and π-dimer, Hdim and Sdim, are summarized in Table 2-2 and Table 2-3.
(a)
(b)
Figure 2-11. (a) Simulated and experimental ESR spectra of 2+• and (b) isotropic Fermi contact couplings for H aroms (|aH| > 0.9 G) calculated at the B3LYP6–31G(d) level.
-2.2 G
-2.2 G -1.2 G
1.0 G
- 20 -
Figure 2-12. Absorption spectra of 2+•SbF6– (105 ± 4 M) at various temperatures, and van’t Hoff plots are shown in the inset.
Figure 2-13. Absorption spectra of 3+•SbF6– (27.8 ± 0.9 M) at various temperatures, and van’t Hoff plots are shown in the inset.
- 21 -
Figure 2-14. Absorption spectra of 4+•SbF6– (30.8 ± 1.0 M) at various temperatures, and van’t Hoff plots are shown in the inset.
Figure 2-15. Absorption spectra of 5+•SbF6– (575 ± 8 M) at various temperatures, and van’t Hoff plots are shown in the inset.
- 22 -
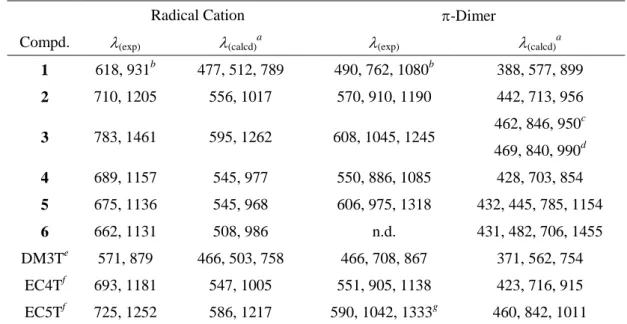
Table 2-2. Observed and Calculated Absorption Bands (/nm) for the Radical Cations and their
-Dimers.
Radical Cation -Dimer
Compd. (exp) (calcd)a (exp) (calcd)a
1 618, 931b 477, 512, 789 490, 762, 1080b 388, 577, 899
2 710, 1205 556, 1017 570, 910, 1190 442, 713, 956
3 783, 1461 595, 1262 608, 1045, 1245 462, 846, 950c
469, 840, 990d
4 689, 1157 545, 977 550, 886, 1085 428, 703, 854
5 675, 1136 545, 968 606, 975, 1318 432, 445, 785, 1154
6 662, 1131 508, 986 n.d. 431, 482, 706, 1455
DM3Te 571, 879 466, 503, 758 466, 708, 867 371, 562, 754
EC4Tf 693, 1181 547, 1005 551, 905, 1138 423, 716, 915
EC5Tf 725, 1252 586, 1217 590, 1042, 1333g 460, 842, 1011
aCalculated at the TD M06-2X/6-31G(d) method. bData from ref. 20. cCalculated with the cccc-conformer. dCalculated with the tcct-conformer. eExperimental values were from ref. 6.
fExperimental values were from ref. 7a. gShoulder band.
Table 2-3. Observed and Calculated Dimerization Enthalpy (Hdim/kcal mol–1) and Entropy (Sdim/cal K–1 mol–1).
Hdim Sdim
Compd. exp calcd exp
1 –6.7a,b –8.9 –27a,b
2 –10.3b –18.5 –31b
3 –12.3b –25.0c
–32b –25.0d
4 –12.4b –20.8 –35b
5 –8.0b –15.4 –29b
6 n.d. –9.3 n.d.
DM3Te –10 –14.0 –25
EC4Tf –13.9 –22.1 – g
EC5Tf –15.5 –27.4 – g
aData from ref. 20. bThe experimental error were within ± 1 kcal mol–1 and ± 6 cal K–1 mol–1.
cCalculated with the cccc-conformer. dCalculated with the tcct-conformer. eExperimental values were from ref. 6. fExperimental values were from ref. 7a. gNot described in ref 7a.
- 23 - DFT Calculations.
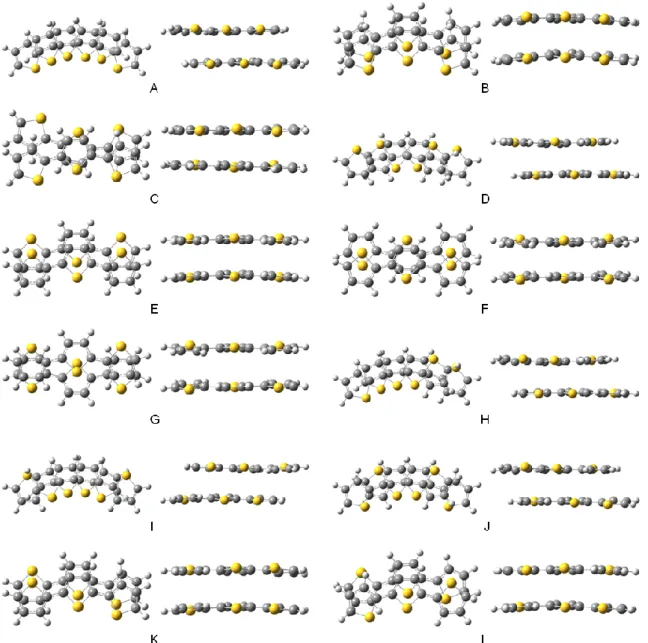
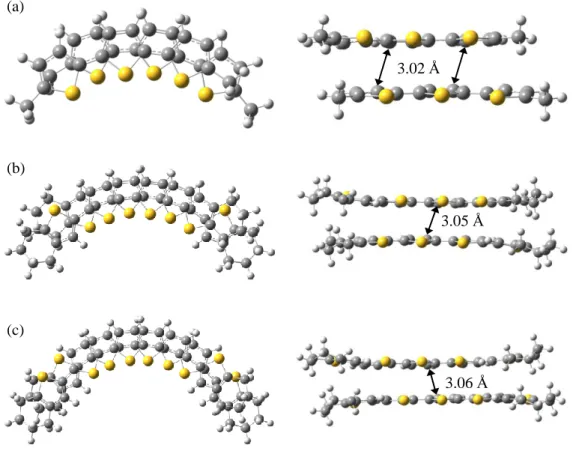
To consider the factors driving the -dimerization of the oligothiophene radical cations, DFT calculations of the -dimers were conducted at the M06-2X/6-31G(d) level.24 First, the various structures of the -dimers of unsubstituted terthiophene (3T) radical cations were optimized in dichloromethane using the polarizable continuum model (PCM). From the twelve possible optimized structures, the face-to-face dimers of cis-cis (cc-) or cis-trans (ct-) conformers with slipped structures along the long axis were found to be more stable than the trans-trans (tt-) conformers (Figure 2-16), which was in contrast to the result that the tt-conformer was the ground-state structure of the monomer radical cation. In the oligothiophene radical cation, the SOMO resides mainly on the
-carbons and less on the sulfur atoms. Thus, these findings are considered to result from maximizing the overlap of the SOMOs of the monomer32 and preventing the steric repulsion between the sulfur atoms. In the dimers of tt-conformers, the overlap of the SOMOs appears to be small in the slipped structure. On the other hand, the spin-unrestricted broken symmetry method33 was performed to check the biradical character of the dication. However, the calculations gave identical states to the spin-restricted method, which indicates that the closed-shell singlet is in the ground-state for (3T)2
2+ at this level of theory. The calculated dimerization enthalpy Hdim(calc) in dichloromethane solution (Table 2-4) for the most stable dimer (–10.1 kcal mol–1) was in close agreement with the experimental value (–10 kcal mol–1),6 but Hdim(calc) in vacuo showed a positive value (+33.9 kcal mol–1). Thus, these methods of calculations suggested that the dimerization in dichloromethane solution was principally driven by the greater stabilization of the -dimer (–113.4 kcal mol–1), which is much greater than that of the monomer radical cation (–34.7 kcal mol–1).
Next, the same calculations for (1)22+
were performed and the most stable structure among the possible optimized conformers was found to be almost identical to the X-ray structure.20 As shown in Figures 2-7, both of the structures were in the cc-conformer with a slipped structure along the long axis, as observed in (3T)2
2+. The nearest intradimer C–C contact was found between the
-position of the central thiophene rings, and the calculated lengths (2.95 Å in dichloromethane, 2.99 Å in vacuo) nearly the same as the observed length (2.98 Å). Such intermolecular interactions that were around 3 Å and caused by the overlap of the SOMOs of the radical species were referred to as
“multicenter two-electron bonds” by Miller and Novoa,1d,3d,4,13b
and these interactions have attracted growing interest. In the case of (3T)2
2+, there was no significant difference in the calculated “bond length” between the optimized structures in vacuo and in dichloromethane. Thus, it is likely that the solvation did not significantly alter the nature of the “bond”, even though the electrostatic interaction with the counter anion plays a critical role to stabilize the -dimer structure in the solid state.3b,c,4
- 24 - Figure 2-16 The optimized structures of (3T)2
2+ in dichloromethane at the M06-2X/6-31G(d) level using the polarizable continuum model (PCM).
Table 2-4. The Calculated Dimerization Enthalpy (Hdim(calc)/kcal mol-1) of (3T)22+ in Dichloromethane Solution at the M06-2X/6-31G(d) Level with the PCM.
Structurea A B C D E F G H I J K L
Hdim(calc) -8.9 -5.0 -3.0 -7.8 -7.2 -5.1 -2.8 -8.2 -10.1 -9.1 -6.1 -5.9
aThe alphabets are corresponding to the alphabets in Figure 2-16.
- 25 -
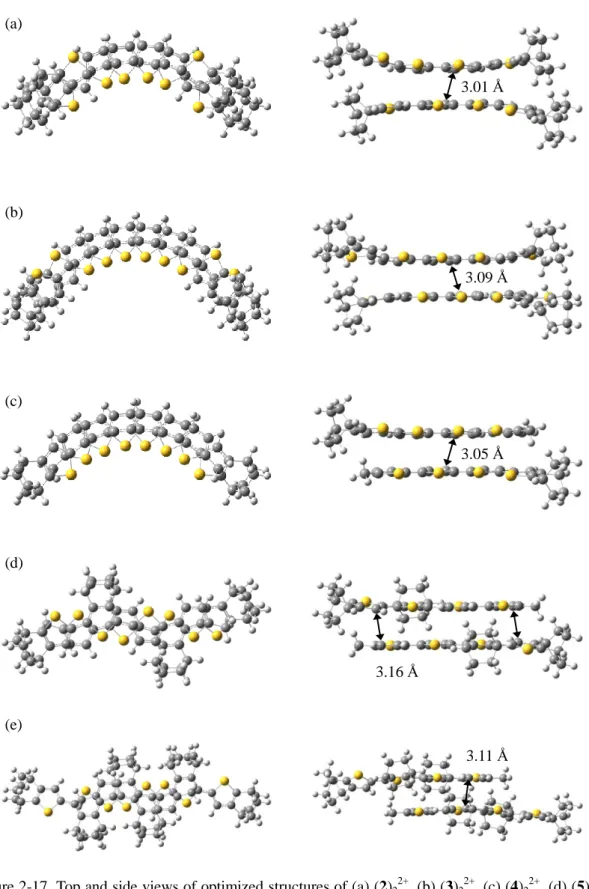
Next, the M06-2X method was applied to the calculations for (2)2 2+– (6)2
2+, together with (DM3T)2
2+ and (ECnT)2
2+, which also showed π-dimerizations in dichloromethane solution.6,7a As shown in Figures 2-17 and 2-18, the most stable structures of all of the π-dimers except for (5)2
2+
and (6)2
2+, showed essentially the same trend as that observed in (3T)2
2+, i.e., the face-to-face dimers of the conformer having all-cis bonds or almost all-cis bonds but with a trans bond at the terminal position with a slipped structure along the long axis. In the calculations of (3)2
2+, nearly the same enthalpy changes were observed in the two conformers having all-cis bonds (cccc-conformer) and almost all-cis with trans bonds at both of the terminal positions (tcct-conformer). Judging from the Time-dependent (TD) DFT calculations34 described below, the tcct-conformer was considered to be the actual structure of (3)2
2+. In contrast, (5)2
2+ and (6)2
2+ displayed an all-trans (ttt-) conformation because of the steric repulsions between the bridgehead protons and -protons in the neighboring thiophene rings or between the bridgehead protons themselves, where less effective overlap of the SOMOs is available. In this context, it is worth noting that the X-ray structure of -dimer of diphenyl terthiophene radical cation with dibutyl substituents at -positions of central thiophene ring was in an all-trans conformation9 probably due to the similar steric repulsion. On the other hand, even the longest (3)22+
gave identical results in both RM06 and UM06 with the broken-symmetry method, which indicates again that no biradical character was observed in this method.
TD-DFT calculations in dichloromethane at the M06-2X/6-31G(d) level were also conducted for all of the π-dimers with the most stable structures to support the assignment of the observed absorption bands. The results of the TD-DFT calculations are summarized in Table 2-2 with the corresponding experimental results. The results of calculations were in close agreement with the experimental values, although the TD-DFT calculations systematically overestimated the transition energies. Regarding the assignment of the longest absorption band, the excitation was dominated by the HOMO-LUMO transition, i.e., the CI expansion coefficients of the HOMO–LUMO transition in the longest absorption bands of all the -dimers were approximately 0.7, and the contribution of the HOMO–LUMO transition was in the range of 96% to 100%. On the other hand, it is notable that the M06-2X method reasonably reproduced the observed difference in the transition energy caused by the structural changes in the -dimer, such as bending in (1)2
2+, and the conformational changes from all-cis in (4)22+
to all-trans in (5)22+
, which again supports the efficiency of these methods. In (3)22+
, two most stable conformers, i.e., the cccc-conformer and tcct-conformer, were obtained, and the calculated values were compared with the experimental values. As a result, although the longest absorption band of (3)22+
observed in the experiments (1245 nm) was longer than that of (2)22+
(1190 nm), the calculated longest absorption wavelength of (3)22+
in the cccc-conformer (950 nm) was shorter than that of (2)22+
(956 nm) in contrast to a longer wavelength in the tcct-conformer (990 nm). Thus, it was concluded that the actual structure of (3)22+
in solution is the tcct-conformer.
- 26 - (a)
(b)
(c)
(d)
(e)
Figure 2-17. Top and side views of optimized structures of (a) (2)22+
, (b) (3)22+
, (c) (4)22+
, (d) (5)22+
, and (e) (6)22+
calculated at the M06-2X/6-31G(d) level using the PCM.
3.01 Å
3.09 Å
3.05 Å
3.16 Å
3.11 Å
- 27 - (a)
(b)
(c)
Figure 2-18. Calculated structures of side and top views of (a) (DM3T)2
2+, (b) (EC4T)2
2+ and (c) (EC5T)2
2+ in dichloromethane at the M06-2X/6-31G(d) level using the PCM.
2-6. Discussion.
As described in the section of the calculations of (3T)2
2+, the dimerization of radical cations in vacuo is enthalpically unfavorable because of the large Coulomb repulsion between the cations. However, the M06-2X with the PCM method showed that the dimerization became an enthalpically favorable process in dichloromethanesolution because the solvation energy of the dimer dications was much higher than twice the solvation energy of the corresponding monomer cations. It is known that such -dimerizations are promoted in more polar solvents, such as acetonitrile,8 and this phenomenon is supported by the greater solvation energy of the -dimer with more polar solvents. However, polar solvents tend to destabilize radical cations in the solution phase, and most studies of -dimer-based supramolecular chemistry are conducted in dichloromethane.7,10,12,15,16 Therefore, to shed some light on the driving forces for the -dimerization in dichloromethane solution, the following four factors were considered: (1) SOMO–SOMO interactions, (2) van der Waals forces, (3) solvation and (4) Coulomb repulsion. Each factor will be discussed in detail.
3.02 Å
3.05 Å
3.06 Å
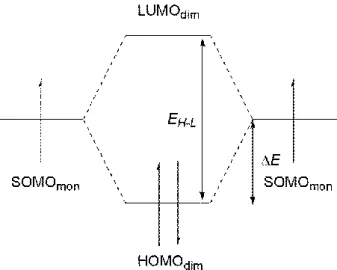
- 28 - SOMO–SOMO Interactions.
Upon -dimerizations of radical cations, the orbital interactions among the SOMOs result in the formation of a pair of bonding and anti-bonding orbitals, which corresponds to the HOMO and LUMO of the -dimer, and two electrons from each SOMO drop into the HOMO. As shown in Figure 2-19, when the HOMO level is stabilized by E from the SOMO level, the total stabilization energy of 2E is thought to be generated upon -dimerization. If it is assumed that the LUMO level is destabilized by E, the stabilization energy 2E can be roughly estimated from the HOMO–LUMO gap of the -dimer i.e., EH-L.21 The estimation of the HOMO–LUMO gap directly from the difference between the Kohn-Sham (KS) HOMO and LUMO levels would be inappropriate, because the values determined with the M06 suits depends on the degree of the Hartree Fock exchange. For example, the differences in the KS HOMO and LUMO levels of (1)22+
were calculated to be 60 kcal mol–1 at the M06-2X/6-31G(d) level and 14 kcal mol–1 in M06-L/6-31G(d).
In contrast, because the excitation energy of the first singlet excited state is a reasonable approximation of the HOMO-LUMO gap,35 the experimental and calculated longest absorption bands were used for EH-L. Then,the energy difference arising from the SOMO–SOMO interaction
ES–S can be expressed in the following Equation (1) because ES–S in the radical cation monomer is null.
ES–S = – EH-L (1)
The results are summarized in Table 2-5. In the comparison between the -dimer of terthiophene (1)22+
bearing a bent structure and (DM3T)22+
with a parallel stack, the absolute values of ES–S for (1)2
2+ (ES–S(exp): –26.5 kcal mol–1, ES–S(calcd): –31.8 kcal mol-1) was lower than those for (DM3T)2
2+(exp: –33.0 kcal mol–1, calcd: –37.9 kcal mol–1). The lower value is thought to arise from the less efficient orbital overlap in (1)22+
,as shown in Figure 2-20. A similar relationship was also observed between (2)22+
and (EC4T)22+
, whereas the opposite trend was observed between (3)22+
and (EC5T)2
2+. Thus, in the -dimers of longer oligothiophenes, such as quinquethiophenes, the effects of the bent structure on the SOMO–SOMO interactions become negligible, most likely because there is sufficient overlap of the SOMOs in the middle part of the -system (Figure 2-21).
Among quaterthiophenes 4-6, (4)2
2+, which is in an all-cis conformer, had the greater ES-S
(exp: –26.3 kcal mol–1, calcd: –33.5 kcal mol–1) than those of (5)22+
(exp: –21.7 kcal mol–1, calcd:
–24.8 kcal mol–1), which is in an all-trans conformer, and (6)22+
(calcd: –19.6 kcal mol–1), which is also in an all-trans conformer with the narrowest contact area. On the basis of these results, it is believed that for the same oligomer length of the shorter oligothiophene radical cations, the efficient overlap of the SOMO, which is made available in a cis bond at the central position with a larger contact area, brings about a higher stabilization energy because of the efficient SOMO–SOMO interaction. In contrast, although the contact area increases with an increase in the number of thiophene rings, the stabilization energy generated by the SOMO–SOMO interaction decreases with
- 29 -
increasing chain length, most likely because the spin density at each interacting site is decreased by the delocalization of the SOMO in the larger -system. As for the calculated Mulliken spin density at the UM06-2X/6-31g(d) level, the sum of the values on carbons in the central thiophene ring were 0.43 for 1+•, 0.35 for 2+•, 0.32 for 3+•, respectively.
Figure 2-19. Schematic illustration of the stabilization by the SOMO–SOMO interaction.
(a) (b)
Figure 2-20. HOMO of (a) (1)22+
and (b) (DM3T)22+
(Isovalue = 0.025).
(a) (b)
Figure 2-21. HOMO of (a) (3)22+
and (b) (EC5T)22+
(Isovalue = 0.02).
- 30 -
Table 2-5. The Calculated -Dimerization Energies (Edim), SOMO–SOMO Interactions (ES-S(calcd)), and Differences in van der Waals Force (EvdW), Solvation Energies (Esol), and Intradimer Coulomb Repulsion Energies (ECoul), together with the Experimental SOMO–SOMO Interactions (ES-S(exp))and Calculated Solvation Energies of Radical Cation Monomer (Esol mon) and -dimer (Esol dim) in kcal mol–1.
Compd. Edim ES–S (exp) ES–S (calcd) EvdW Esol mon Esol dim Esol ECoul
1 –10.0 –26.5a –31.8 –1.8 –28.6 –93.5 –36.3 60.0
2 –19.7 –24.0 –29.9 –7.9 –27.0 –88.0 –33.9 52.0
3 –26.4 –23.0 –28.9 –10.7 –26.3 –84.0 –31.3 44.8
4 –21.3 –26.3 –33.5 –4.3 –28.4 –92.8 –36.0 52.4
5 –17.3 –21.7 –24.8 –12.6 –26.9 –86.7 –32.8 52.9
6 –10.3 n.d. –19.6 –10.0 –25.2 –81.0 –30.6 49.9
DM3Tb –14.2 –33.0 –37.9 –0.1 –32.1 –106.5 –42.2 66.0
EC4Tc –23.7 –25.1 –31.2 –10.0 –27.7 –91.7 –36.3 53.8
EC5Tc –29.9 –21.4d –28.3 –15.4 –27.1 –87.2 –32.9 46.7
aData from ref. 20. bExperimental value in ref. 6 was used. cExperimental values in ref. 7a were used.
dCalculated from shoulder peak.
Van der Waals Forces.
In the radical cation -dimer, van der Waals forces, as observed in the interaction between neutral molecules, should also play a role. It was assumed that such a change in intermolecular van der Waals forces EvdW from the monomer radical cation to the radical cation -dimer, excluding the effect of the SOMO–SOMO interactions and Coulomb repulsion, can be estimated using the total energies obtained from the single-point calculations of the neutral molecule at the radical cation and
-dimer geometry, i.e., En@rc and En@pd, respectively. Thus, EvdW was calculated by the subtraction of twice the En@rc from En@pd [Equation (2)]:
EvdW = En@pd – 2× En@rc (2)
Regarding the structure of the -dimers, (1) 2
2+ has a bent structure because of the steric repulsion of BCO groups and hence has a narrower contact -area than (DM3T)2
2+, which has a parallel stack. In the comparison of EvdW values (Table 2-5), structural differences were shown to have little influence. Thus, the repulsive steric effect appears to be countered by attractive van der Waals forces between the BCO groups and the opposite -surface (CH…interactions). However, ECnT, which has less bulky cyclohexene units, demonstrated higher EvdW values than both 2 and 3, respectively, most likely because of the additional van der Waals interactions between the larger cyclohexene units as compared with the methyl groups.
- 31 -
The term EvdW involves the energy difference generated by the conformational change from all-trans in the radical cation monomer to the preferable mainly cis-connected banana-shaped conformer of the -dimers. Because it is known that neutral oligothiophenes also prefer the trans conformer to the cis conformer,36 EvdW for -dimers having banana-shaped conformers such as 1-4, includes some energy loss because of the change in the disfavored cis conformer, which was shown to be overcome by the more effective SOMO–SOMO interactions. Thus, EvdW for 5 and 6 (–12.6 and –10.0 kcal mol–1) in the all-trans conformer for the -dimer is higher than that for 4 (–4.3 kcal mol–1) by 8.3 kcal mol-1, whereas ES–S for 4 (–33.5 kcal mol–1) is higher than that for 5 (–24.8 kcal mol–1) by 8.7 kcal mol-1 (see Table 2-5).
Regarding the chain length dependence, EvdW increased with increasing chain length.
Judging from the additivity of van der Waals forces and the calculated interaction energy between neutral thiophene molecules (2-3 kcal mol–1),37 the difference in EvdW from EC4T to EC5T (5.4 kcal mol–1) may be somewhat overestimated.
Solvation.
In the Born model of ion-solvation,38 the free energy change can be expressed by Equation (3):
(3)
where Q, and represent the Born excess free energy of solvation, the ionic charge, the radius and the solvent dielectric constant, respectively. From this equation, it is obvious that the free energy change increases with increasing ionic charge Q and with decreasing ion radius . In other words, the ion-solvent interaction becomes stronger as the charge concentration becomes denser and as the size becomes smaller. Thus, when radical cation monomers form -dimers, the solvation energy increases because of the denser charge concentration in the -dimer. At the M06-2X/6-31G(d) level with the PCM, the difference in the solvation energy between the radical cation and -dimer was found to be the important driving force for the -dimerization of oligothiophene radical cations even in dichloromethane, which is a moderately polar solvent. As described above, the calculations for
-dimers in vacuo showed that -dimerizations are unfavorable without solvation. Thus, the solvation energies of the monomer Esol mon were estimated from the differences in the calculated total energy of radical cation monomers in vacuo and in dichloromethane. In the same manner, the solvation energies of the -dimer Esol dim were estimated. Then, the stabilization energies upon
-dimerization by solvation, i.e., Esol, were estimated from Equation (4):
Esol = Esol dim – 2Esol mon (4)
The results for Esol, as well as Esol mon and Esol dim, are also summarized in Table 2-5.
- 32 -
The Esol mon value ranged from –25 to –32 kcal mol–1, and the stabilization energy increased with decreasing molecular size, which gave an energy difference of 7 kcal mol-1 from DM3T to 6. The Esol dim value also showed a similar change, but the range of stabilization energy (–81 to –107 kcal mol–1) and the energy difference (26 kcal mol–1) were much higher than the in the Esol mon values because of the denser charge concentration in the dication (i.e., the dimer of the monocation). As a result, Esol was larger for the smaller molecules including their substituents. For example, among 1-3, DM3T and ECnT, the Esol value decreased with increasing chain length.
Additionally, among quaterthiophenes 2, 4-6 and EC4T, the Esol value decreased with increasing size of the substituents, which indicates that the charge delocalization into the BCO units decreased the charge concentration in the -systems.
Coulomb Repulsion.
The three factors mentioned above, i.e., ES–S, EvdW, and Esol, are driving forces for the
-dimerization of radical cations, whereas Coulomb repulsions should greatly suppress the
-dimerization because of the intermolecular interactions between the positively charged species. If it is assumed that these four factors dominate the -dimerization process, the change in the
-dimerization energy Edim can be expressed as Equation (5):
Edim =ES-S + EvdW + Esol + ECoul (5)
where Edim was used instead of Hdim,ES–S, EvdW, Esol, were described above, and ECoul was the energy of Coulomb repulsion. Because Edim, ES–S, EvdW, and Esol were already obtained,
ECoul was simply calculated from Equation (5), and the values are also summarized in Table 2-5.
In the comparison among the molecules included under 1-3, the ECoul value decreased with increasing chain length, and the same trend was also observed for DM3T and ECnT.
Furthermore, the ECoul value for 4-6 decreased with an increase in the number of BCO units.
Similarly, the ECoul value for 1 was found to be lower than the ECoul value of DM3T. These results are consistent with the notion that the Coulomb repulsion was weakened by delocalization of the positive charge in the larger -system and electron-donating BCO groups. In contrast, the ECoul
values for 2 and 3 were almost identical to the ECoul of EC4T and EC5T, respectively, which suggests that cyclohexene units and BCO groups have a similar electron-donating character.
The effect of Chain Lengths and Structures of the π-Dimer on Hdim.
As for the calculated -dimerization enthalpy Hdim(calcd) in Table 2-3 (the values nearly equal to Edim in Table 2-5), the absolute value increased with increasing chain length in the order of 1 > 2 > 3 and DM3T > EC4T > EC5T. These results were in reasonable agreement with the