and Their Application to an Organic Secondary Battery
A Thesis Presented to Waseda University
Waseda University
Graduate School of Advanced Science and Engineering Department of Applied Chemistry
Research on Polymer Chemistry
February 2013
Il Seok CHAE
Referees: Prof. Dr. Takayuki Homma Prof. Dr. Kenichi Oyaizu Prof. Dr. R. C. Advincula
The history of polymer electrode for batteries is as old as that of inorganic ones. In fact, the first commercialized primary lithium battery adopted the (CF)n– Li configuration in 1973. Since the late 1970’s with the discovery of the electric conductivity of doped polyacetylene, there was a lot of enthusiasm about flexible lithium/polymer cells that use Intrinsically Conducting Polymers (ICPs). Nevertheless, with the tremendous success of inorganic intercalation compounds in the late 1980’s, and with the development progress of ICPs as rechargeable cathodes being slow, interests on organic electrodes have decreased. This was mainly due to the insufficient doping level of ICPs. Consequently, the redox capacity was low, and the doped states were not chemically stable, which have lead to self-discharge and degradation of the rechargeable properties of lithium/ICPs cell.
On the other hand, in inorganic battery, the lithium-ion battery, first commercialized by Sony in 1991, has been emerged from the principle of the Li+ exchange between the graphite anode and a layered-oxide cathode. This ion transport phenomenon was firstly proposed by Armand and Scrosati, who named them
“rocking-chair” batteries. Other names are “SWING”, “shuttlecock”, “Li-ion”, or “Lion cells”. Thereafter, the battery-science community has found out the other problem in cathode-active materials based on organic polymer.
The redox process of most of ICPs is associated with the electroneutralization of anions (X–). The is due to that most of ICPs work as p-type charge storage behaviors. Also, the charge-storage of n-type redox activity of the ICPs still has been limited. In lithium/ICPs cell, the anion migrates to the ICPs electrode, and the Li+ migrates to the Li metal during the charging process. Consequently, the salt concentration in the electrolyte changes during charging/discharging cycling. Therefore, enough solvent must be provided to leave the salt LiX dissolved. This is unfavorable for the specific/volumetric energy density and operation of the battery.
This thesis is concerned with the studies of ‘‘rocking-chair” type cation migration by synthesis of the zwitterionic redox-active radical polymers, which are inspired by the self-doped conducting polymers.
Although the n-type redox active radical polymers are already demonstrated as a ‘‘rocking-chair” battery, they show relatively lower performances than p-type radical polymers. Therefore, the development of p-type radical polymers for a ‘‘rocking-chair” type battery design is an urgent requirement for compact energy storage devices. Chapter 1 introduces the radical polymer battery, the self-doped conducting polymer and the carrier-mediated mass transfer. Chapter 2 describes the synthesis of zwitterionic radical polymers bearing vinyl sulfonic acid and the investigation of ion tranport phenomena in aqueous electrolyte. Chapter 3 reports the novel Li-ion radical battery via utilizing styrenesulfonate as a Li+ host material. The effects of zwitterionic configuration and conformation on the redox behavior are described in Chapter 4. Finally, Chapters 5 and 6 discuss the permselective facilitated mass transfer via the fixed carrier silver nanoparticles with organic electron acceptor and the energetics at metal/organic interface. Conclusions and future prospects are given in Chapter 7.
Il Seok Chae
CONTENTS
Preface
Chapter 1 General Introduction
1.1 General Introduction …..5
1.2 Polymers and Rechargeable Batteries …..6
1.3 Electroactive Polymer with Fixed Anionic Groups …..17
1.4 Facilitated transport Phenomena …..25
References …..28
Chapter 2 Self-doping Inspired Zwitterionic Pendant Design of Radical
Polymers toward a Rocking-chair-type Organic Cathode-active Material
2.1 Introduction …..33
2.2 Preparation of Poly(TEMPO-methacrylate-stat-vinyl sulfonic acid) …..35 2.3 The electrochemical properties involving cation migration …..40
2.4 Experimental Section …..43
References …..50
Chapter 3 Efficient Redox Equilibrium of the Zwitterionic Radical Polymer in Non-aqueous Electrolyte for Novel Li+ host material
in a Li-ion Battery
3.1 Introduction …..53
3.2 Preparation of Copolymer …..54
3.3 Electrochemistry of the Radical Polymer Bearing
Styrenesulfonate Group in Non-aqueous Medium …..55
3.4 Experimental Section …..58
References …..62
4.1 Introduction …..63 4.2 Synthesis of Random and Block Copolymers …..64 4.3 Redox Behaviors Depending on Copolymer Configurations …..65 4.4 Copolymer Conformations Effect on Redox Capacities …..67
4.5 Experimental Section …..70
References …..72
Chapter 5 Surface energy level tuning of silver nanoparticle for facilitated olefin transport
5.1 Introduction …..73
5.2 Results and Discussion …..73
5.3 Experimental Section …..78
References …..82
Chapter 6 Highly Polarized Anatase TiO2 Nanoparticles by Poly(ethylene phthalate)
6.1 Introduction …..83
6.2 Results and Discussion …..85
6.3 Experimental Section …..87
References …..88
Chapter 7 Future Prospects
7.1 Conclusions …..89
7.2 Future prospects …..91
References …..93
List of publications Acknowledgement
Introduction
1.1 General Introduction
1.2 Radical Polymers and Rechargeable Batteries 1.3 Electroactive Polymer with Fixed Anionic Groups 1.4 Facilitated transport Phenomena
References
1. 1 General Introduction
Polymers have emerged as one of the most important electro-active material in the twenty-first century considering that constant global energy demand should be fulfilled in a sustainable way. In parallel, new challenges for lithium batteries are opening up in green energy.1 Up to date, a variety of electrodes in lithium-ion batteries, such as LiCoO2 and LiMn2O4, are not produced from renewable energy resource. They are produced from ores, consuming a large amount of energy to extract the raw materials. For example, the cobalt must be obtained from natural resources which make up 20 parts per million of Earths ores;2 consequently, manufacturing the electrodes require considerable energy amount. Therefore, there have been the tremendous efforts to develop the electro-active organic molecules as cathode materials composed of intrinsically conducting polymers (ICPs), but the results have been disappointing.3This was mainly due to the insufficient doping level of ICPs.4
Fortunately, the problems of ICPs have been solved out since the emergence of a new strategy for electroactive organic polymers based on the integration of singly occupied molecular orbitals of radicals as the carriers of hopping electrons.5 This is so-called radical polymers, which are non-conjugated polymers bearing organic robust radicals, such as NO-centered nitroxides, and O-centered phenoxyls and galvinoxyls, as pendant groups per repeating unit. The principle of charge transfer and storage by radical polymer is totally different from those by -conjugated orbitals of ICPs. The organic robust radicals (R) show their electrochemically reversible redox properties. For example, a neutral R is oxidized to a cation, R+(p-type) and reduced to an anion, R by one-electron reactions (n-type). With the redox from R to R, in particular, poly(galvinoxylstyrene) has been demonstrated as a
rocking-chair type battery,6where polymer served as a Li+-inserting cathode as well as conventional lithium metal-oxide-based cathodes. However, the redox potential of n-type is generally lower than the one of p-type.
In this thesis, the author tried to develop the rocking-chair type cathode-active materials, via the polymers bearing the TEMPO and sulfonate anionic groups.7This strategy was inspired from the self-doped conducting polymer which has ionic groups. The principle is that the fixed anionic groups lead to charge compensation of R+ during the one-electron oxidation of the R, i.e., switching to the zwitterionic configuration. The main interest is associated with not only the redox behaviors of radicals, but also the effects of the anionic
moiety bound to polymer backbone.
The remaining sections of this chapter offer a short review of the exisiting literature of radical polymers and the self-doped conducting polymer. Also, the facilitated transport phenomena are described with a veiw of the permselective mass transport via the fixed carrier.
1.2 Radical Polymers and Rechargeable Batteries
1.2.1 Introduction
Organic radicals bearing polymers is bringing us closer to solving the problems of organic based rechargeable batteries.5-7 When, in the 1980s, Bridgestone-Seiko and VARTA/BASF launched commercial cells based on ICPs, such as poly(pyrrole) and poly(aniline).8Nevertheless, there were a lot of challenge toward the polymer material for the next generation of batteries, ICPs have still suffer from limitations in terms of maximum doping density/capacity, chemical stability/cyclability, and rate capability. On the other hand, in 2002 Nakahara et al. first reported a battery using a poly(methacrylate) bearing the persistent 2,2,6,6-tetramethyl piperidinyloxyl (TEMPO) radical as active cathode material (PTMA).9 The cells fabricated with radical polymers display striking battery performances, like fast charging times, stable cell voltage and flexible batteries. These reasons are due to a totally different concept from ICPs, i.e., -conjugated polymers as the organic molecules for electronic devices. In this section, the redox properties of nitroxide radicals, the working principles and the characteristics of radical polymer battery are described with a view of challenging toward rechargeable batteries.
1.2.2 Stable Nitroxide Radicals
Stable radicals are persistent under ambient conditions, even after incorporation into polymers as the pendant group per repeating unit of the backbone. This exceptionally robust property of radical imposes stable battery performance, such as a constant cell voltage, coulombic efficiency, cyclic ability and chemical stability, on the radical battery.
History of Nitroxide Radicals
In 1901, Piloty and Schwerin prepared the first organic nitroxide, porphyrexide,10 and after the emergence of Electron Paramagnetic Resonance (EPR) spectroscopy, its radical character was elucidated by Holden et al.11 The next important contribution to the development of nitroxide chemistry came from Wieland et al.12 and Meyer et al.,13 who prepared diarylnitroxides. The Lebedev et al.14 firstly reported the synthesis of 2,2,6,6-tetramethyl-4-piperidone-1-oxyl (4-oxo-TEMPO), in 1959. Rozantsev et al.14 demonstrated that nitroxide radicals can be involved in many kinds of organic reactions without involvement of the aminoxyl radical. Over two decades in 19601980, the fundamental physicochemical properties of robust nitroxides were also established.14-15
N NH HN
NH O
N O O
MgBr + N
O N
OMgBr
1) H3O+
2) Ag2O N O (Wielandet al.)
NH
OMe
MeO PhCO3H
N
OMe MeO
O (Meyeret al.) porphyrexide
(Piloty and Schwerin) 4-oxo-TEMPO (Lebedevet al.)
Figure 1.2.1The pioneering research on stable nitroxide radicals.
Redox of Nitroxide Radicals
The stability of the NO group allowed chemical reactions of the nitroxides which do not involve the radical center allowing further nitroxyl radicals derivitizations. A nitroxide can be reversibly n-doped to aminoxy anion and p-doped to oxoammonium cation in cathodic
and anodic reactions, respectively. On the other hands, the aminoxyl group of nitroxides is characterized by a N O three-electron bond resulting from the overlap of the 2pz orbitals of the nitrogen and oxygen atoms. The contribution of the two main mesomeric structure in terms of valence bond theory is shown in Figure 1.2.2, suggesting the actual electronic structure. The bond energy is changeable depending on the structures. As a result of this odd-numbered electron in the NO system, the NO bond of an aminoxyl group has a bond order of 1.5, as indicated by ca. 100 kcal/mol of the bond energy, and the bond length dNO
(1.25 Å<dNO <1.30 Å) is midway between the energy and the bond length of a NOH single bond (53 kcal/mol, ~1.43 Å ) and a N=O double bond (145 kcal/mol, ~1.20 Å).16-18
N O
N -e- O
+e- N O
X
N O nitroxide radical oxoammonium cation
(a) (b)
a nitroxide a nitronyl nitroxide Figure 1.2.2 (a) The Redox couple of a nitroxide radical and oxoammonium cation, and (b) mesomeric structures of a nitroxide and a nitronyl nitroxide.
The most important features of the redox reactions of nitroxides are its remarkably rapid and reversible one-electron transfer for the oxidation process. Using nitroxides with aliphatic and aromatic substitutes, their kinetic studies in solutions showed electron-transfer rate constants in the order of 10 1 cm s 1.19 These values are even higher than that of ferrocene (~10 2 cm s 1), a standard molecule used as an internal calibrator in electrochemistry. Table 1.2.1 shows the electron-transfer rate constant (k0) of redox species. The electron-transfer reactions of the radical molecules are almost comparable to that for the copper ion.
Table 1.2.1Electron-transfer rate constant (k0) of redox species19
X-ray Structures of Nitroxide Radicals
X-ray diffraction studies of some representative cyclic nitroxides, (five-, six- and seven-membered rings) reported that the NO bond length is not significantly influenced by the ring size.20 Figure 1.2.3 shows the crystallographic structures data of the nitroxide of six-membered rings, i.e., TEMPO derivative and its oxoammonium compound.21 Due to the sp2 character of the nitrogen atom, the oxoammonium cation was characterized by the double bond nature of N-O and almost planar C2O plane. However, the structures of the radical and its cation were only slightly changed from each other, which had an important role on the electron-transfer reaction of the radical molecule.
Figure 1.2.3Structural implications of TEMPO/TEMPO+BF4 21
An account for the rapid electrode reaction can be attributed by the relatively small atomic rearrangement required for the overall redox reaction to decrease the activation energy, whereas a large rearrangement of structural framework during the redox reactions through the bond scission and formation that must overcome activation energies.
Electron Transfer of the Nitroxide Radical Polymers
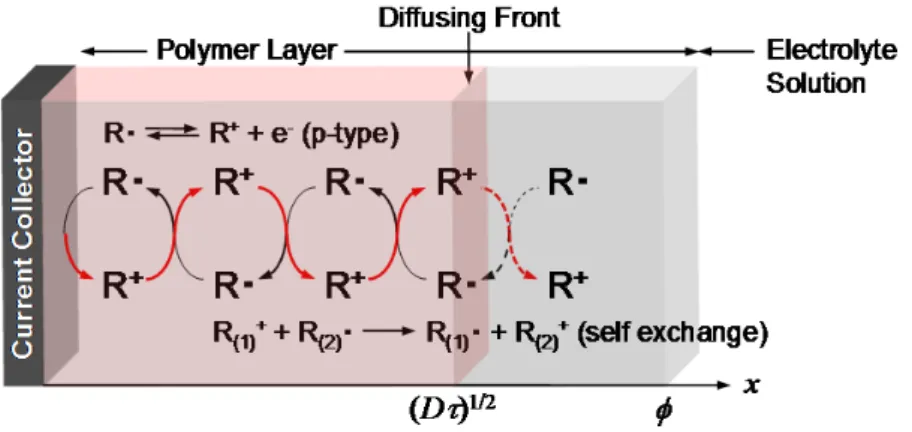
The electrode of the radical polymer battery was prepared by coating the polymer on a current collector, such as ITO, FTO and glassy carbon substrate. In the coated radical polymer layer, a charge is transferred (or propagated) between the neighboring radical moieties by an electron-hopping mechanism during the redox process of the polymer. Also, the redox process involved the transfer of a counter ion (typically an anion) to neutralize the positive charge on the oxoammonium cation generated from the TEMPO radical. Both electron and ion transfer would be the rate-determing step of the charge propagation. It was reported that the diffusion coefficient (D) of the electron- or the charge-propagation process in the radical polymer layer was on the order of 10-9 cm2/s. This value was comparable to those of previously reported redox-active polymers, such as poly(vinylferrocene) (D= 10-9cm2/s).22 The electron transfer process of the nitroxide radical polymer layer confined at a current collector surface was also characterized by normal pulse voltammetry, the results of which suggested a rapid charge propagation (apparent diffusion coefficient,Dapp ~ 10 10cm2/s) throughout the polymer layer in a micrometer scale.
Figure 1.2.4Positive charge propagation during the oxidation of p-type radical polymers confined at a current collector surface and equilibrated in electrolyte solutions, showing the direction of the charge transport in the slab of the polymer.
Nernstian adsorbate-like behaviors were also observed in the case of tens and hundreds of nanometers thick TEMPO-based radical polymer layer.22At a layer thickness of 50 nm, a negligible or very small cathodic-to-anodic peak separation of 5 mV was observed. However, when the thickness was increased to 100 and 230 nm, larger peak separation values of 8 and 22 mV were observed, respectively. The peak-to-peak separation was obviously affected by the diffusional process of the propagating charge throughout the layer. Considering their insulating nature of the matrix, the electron transfer process seemed to be dominated by the electron self-exchange reactions of the neighboring redox sites, like a hopping mechanism.
The amorphous, solvated, and slightly swollen structure of the radical polymer ensures good counterion mobility during the electrode process. Based on such a rapid and simple one-electron transfer process, nitroxides are applicable for an electrode active and charge storage material, promising high power rate performance of the battery.
1.2.3 Radical Polymer Batteries
The battery performance, such as cell voltage, specific capacity, coulombic efficiency, cyclic ability and chemical stability, are almost derived from a combination of electrode-active materials. In this section, the radical polymer battery characteristics are evaluated.
Rate Performance
The most interesting feature of the radical polymer battery utilizing radical polymers as the electrode-active materials is the unprecedented high chargingdischarging rate. Due to the fast kinetics and the electrochemical reversibility for the one-electron redox reaction of the TEMPO free radical, the radical polymer battery is capable of exhibiting excellent rate performances. The rate performances are often represented by using a C-rate, which is defined as a current density that is required to fully charge the battery in a given period of time; thus 1C corresponds full charging in 1 h. In the case of the radical battery, charging times are reduced to a few minutes or even seconds. Conventional batteries are relatively slowly charged at 0.1 - 0.5C rates. In this case, it takes several hours for the full-charging. However, due to the fast kinetics of the one-electron redox reaction of TEMPO in the radical polymer battery, charging times are much reduced. The origin of the fast kinetics is strongly related to the structural feature of the TEMPO radical as described in the above section. Such kinetic features are in sharp contrast to those of other organic redox-active molecules based on conjugated -systems that typically suffer a significant structural rearrangement upon the redox reaction.
Cell Voltage
A radical polymer battery exhibits a nearly constant plateau voltage upon discharging.
This is of great advantage in comparison to organic electrodes made of conductive polymers which often show a sloping curve, i.e., a varying cell voltage. TEMPO-based PTMA reveals a redox potential comparable to currently used metal oxides (3.6 V in LiCoO2).9Therefore, the battery consisted of PTMA and lithium as cathode and anode, respectively, promises to have environmental and economical advantages. When using a lithium and a zinc anode, the cell voltages of 3.5 and 1.7 V were achieved, respectively.22-23 Also, the redox potentials of the organic electrode-active materials are tunable, depending on the substituents of the redox-active species. Electron-withdrawing groups (e.g. NO2and CF3) usually bring about the positive shift of the potential to enhance the cell voltage, and electron-donating groups (e.g.
OMe andt-Bu) shift the potential to the negative side to reduce the cell voltage.24
Specific Capactiy
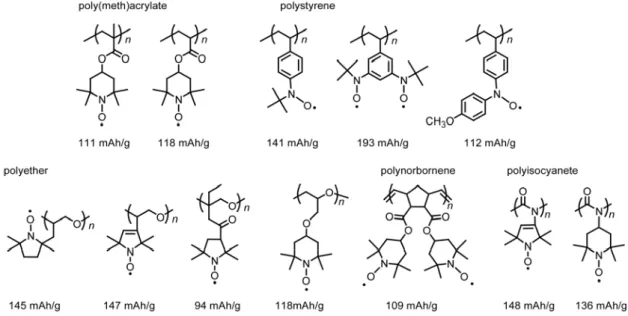
The formula weight-based redox capacity,i.e., the theoretical capacity, is determined by the number of electrons that can be stored per repeating unit/monomer and the molar mass of the one repeating unit of radical polymers. Consequently, the smaller of repeating unit means the higher theoretical capacity. The theoretical capacity of the PTMA, 1 is 111 mAh g 1.9 The lighter poly(vinylether) bearing TEMPO,2 achieved up to 131 mAh g 1.25-26The highest value was achieved with a poly(ethylene glycol) bearing the five membered ring, i.e., PROXYL radical3, 147 mAh g 1.27
N O
O O
N O O
O
N O
n n n
111 mAh/g1 2
131 mAh/g 3
147 mAh/g
Figure 1.2.5Structure of radical polymer bearing TEMPO or PROXYL.
Recently, two-electron oxidation in TEMPO was reported by using for graphene, cellulose and DNA scaffolds, suggesting the increase in capacity. Ideal structures, which are up to now not synthetically available, feature a theoretical capacity of 224 mAh g 1.28 Since the radical polymers have the poor conductivity, conductive agents and binders, which need to be added in fabricating practical electrodes. The electrode yields a specific capacity of 110 mAh g 1in the presence of only 45% of VGCF, approaching the theoretical value of 111 mAh g 1.29Therefore, 55 mAh g 1is reasonable for the capacity of PTMA with the consideration of weight fraction in the electrode.
Cyclic Ability
Literature examples prove a very good cycle lifeexceeding 1000 recharge cycles in many systems. This cyclic ability is mostly due to the chemical stability of the nitroxide radical and the fast kinetics for the one-electron transfer process. As already mentioned in the previous section, almost no significant structural change occurs during the one-electron transfer
reaction. A negligible decrease of capacity was observed during the cycling, which was most likely due to the elution or agglomeration of some part of the swollen polymer.
Chemical Stability and Coulombic Efficiency
Radical polymer battery will meet the expectation of several service life due to the exceptionally robust stability of TEMPO. However, the oxidated state of TEMPO, i.e., the complex of N- oxoammonium cation and anion, is vulnerable to high temperature. The coulombic efficiency is reported in the range of 95% to 97%.
1.2.4 Molecular Engineering of Radical Polymers
Designing multiple radical sites in a repeating unit of a polymer is an effective strategy to increase the theoretical capacity. For example, 4 was proposed to bear two nitroxyl moieties per unit in Figure 1.2.6.30However,4showed the unstable or less electrochemically reversible redox property. The spirobinitroxide polymer 5 bearing the two radical sites was also suggested with a view to accomplish a the theoretical capacity of 174 mAh g 1. Unfortunately, 5 also suffered from the decomposition of the polyacetylene backbone during the oxidation process.31
N N
O O
n
N N
N O
O
O n
4
194 mAh/g 5
174 mAh/g
Figure 1.2.6Structure of radical polymer with the multiple redox center.
A series of radical polymers have been successfully synthesized. Figures 1.2.7 and 1.2.8 show the radical polymers developed by our group. Various polymer backbones, for example, poly(meth)acrylates, polystyrene, poly(vinyl ether)s, polyethers, and poly(norbornene)s, have been employed to bear the radical pendants. The remarkable point is the backbones with a lower glass transition temperatures or rubbery characteristics that often produced a higher rate performance during the charging and discharging processes. In the case of n-type redox active
radical polymers, an electro-withdrawing group, such as a carbonyl and trifluorometyl group, imposes on stabilize the n-type redox pair, also the tunable redox potential property. Thus, the molecular design and synthesis are still effective and powerful routes for developing the new organic functional materials including the organic radical polymers.
Figure 1.2.7 Nitroxide radical polymers based on various polymer backbones
Figure 1.2.8 n-Type radical polymers
1.2.5 Ionic Transport in Radical Polymer Batteries
The conventional Li-ion battery is based on the rocking-chair type intercalation reactions of Li-ions between the cathode and anode materials. During the charging/discharging process, Li+ exchange between the graphite anode and a layered-oxide cathode. On the other hand, the ion transport of radical polymer battery is depended on the redox types.
Figure 1.2.9The ion transport in p-type radical polymer battery during charging/discharging process
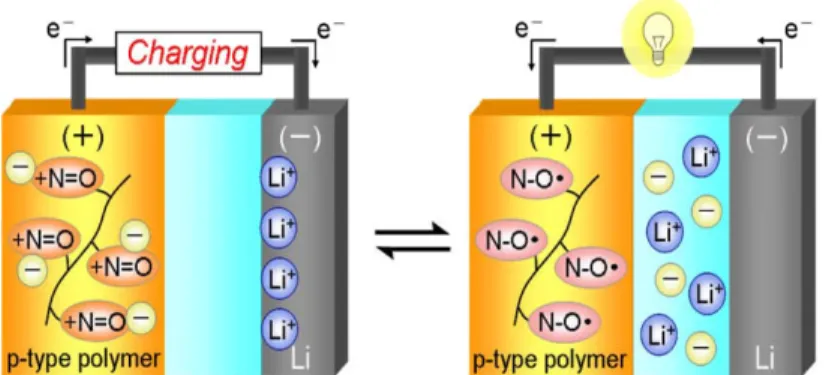
The configuration of p-type radical polymer battery is illustrated in Figure 1.2.9. The cathode is PTNB or PTMA, and the anode is lithium metal. During the one-electron oxidation of the nitroxide radical, anions of Li salt in electrolyte are incorporated into N-oxoammonium cation, and Li+ goes Li metal for the charge compensation. Consequently, the salt concentration in the electrolyte changes during charging/discharging cycling, and the excessive amount of lithium salt is unavoidable to maintain the capacity of electrode and the ionic conductivity in electrolyte
Figure 1.2.10 The rocking-chair type migration of cation in n-type radical polymer battery during charging/discharging process
Due to the inherent n-type redox mechanism, the battery is working on the principle of
rocking-chair type cation migration as shown in Figure 1.2.10.6Thus, only cations were exchanged at both electrodes during charge/discharge cycles, giving rise to the ultimate reduction of the electrolyte down to the minimum amount necessary to allow current flow and to the downsize in overall battery volume. Considering the intrinsically lower volumetric energy densities of organic material, the rocking-chair migration is one of the most ugent problems in organic radical polymer battery.
1.3 Electroactive Polymer with Fixed Anionic Groups
1.3.1 Introduction
Redox reactions are accompanied by not only energy changes in form of heat, light or electricity, but also redistribution of electrons among the reacting species and the migration of ion-species. In particularly, the incorporated of ion-species provide a powerful additional element to determine the electrochemical properties in ICP system, i.e., p-type doping and n-type doping process like follows.
Originally, the conjugated polymers in their neutral state are semiconductors with band gaps ranging from 1 to several eV. In order to make them electronically conductive, it is necessary to introduce carriers into the conjugated system. This process is called doping, which is insertion of conterions resulting from oxidation or reduction. To explain the electronic phenomena in ICPs, concepts of charge carriers such as solitons, polarons and bipolarons, are necessary. Here, the author does not put much emphasis on these terminologies in the field of solid physics.
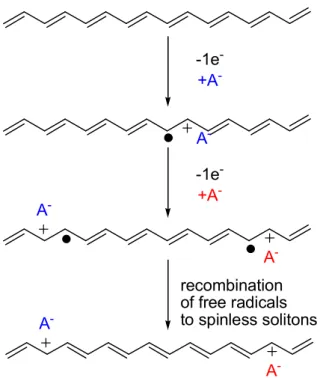
In polyacetylene, the p-type doping is achieved by the chemical or anodic oxidation, resulting in the generation of carbonium cations and radicals. To accomplish the charge balance, equivalent numbers of anions insert into the polymer as shown in Figure 1.3.1.
Contrary to a conventional semiconductor based on silicon, ICPs require a large amount of
dopants. This doping degree not only increases electronic conductivity of ICP, but also induces desired properties such as improved processability, environmental stability and special catalytic, optical and redox properties.
Taking these advantages, ICPs are very attractive materials with potential application to organic devices such as rechargeable batteries, electrochromic displays78,79for smart windows, and light-emitting diodes. Also, ICPs bearing ionic functional groups, i.e., self-doped polymers, are most powerful candidates among the conducting polymers, which are expected to play important roles by overcoming many of the limitations in the conducting polymers without ionic functional groups.
-1e- +A-
A- -1e- +A- A-
A- recombination of free radicals to spinless solitons A-
A-
Figure 1.3.1p-Type doping in polyacetylene (fromProgress in Polymer Science,27, 135.)32
Among the doping process, the doping anion strongly influences the morphology of the polymer. For example, the synthesis of polypyrrole (PPy) via electrochemical oxidation of pyrrole monomers is a well-known and well-controlled method. Anions of different sizes can be used to dope PPy during the polymerization process. Examples of these anions include (1) small anions, such as BF4 , PF6 , ClO4 , NO3 , Cl , and etc; (2) medium-sized anions, such as p-toluensulfonate or benzenesulfonate; (3) huge anions, such as poly(styrene sulfonate) (PSS) or poly(vinyl sulfonate) (PVS). The resulting polymers exhibit different conductive, mechanical, or electrochemical properties depending on the incorporated anion.33-36 Given the conduction mechanism of ICP i.e., solitons, polarons and bipolarons affected by the configuration of polymers, the study of doping anion could give insight into the
physicochemical nature of polymer bearing the fixed anionic group.
Radical polymers have the localized redox centers and the different conduction mechanism as already mentioned in 1.2 section. However, the incorporated anion/cation during redox process is necessary for the electroneutrality in a system. As a strategy of
rocking-chair-type migration of cation, the zwitterionically designed radical polymers were suggested in this thesis, which originally inspired from the concept of self-doping. In this section, the author refer to the book about self-doped conducting polymers by Freund and Diore which was published in 2007,37 and summarized the some feature of self-doped ICPs on the introductory level.
1.3.2 Self-doped ICPs
Emergence of Self-doped Conducting Polymers
In 1987, Wudl and Heeger et al. firstly reported the novel concept of self-doping in conducting polymers as shown in Figure 1.3.2.38 Simply, a self-doped conducting polymer could be described as a conjugated polymer bearing ionic group acting as a dopant anion/cation. This strategy is particularly interesting and should be noted, because ICPs bearing ionic group, by itself, maintain the charge neutrality during the charge injection process.
S
(CH2)nX-M
n S
(CH2)nX- n -e-, -M+
+e-, +M+ M = H, Na, Lietc.
X = SO3
Figure 1.3.2Redox reactions of polythiophene bearing sulfonic group38
The Restricted Role of Self-doping
Using the terminology self-doped need particular attention to avoid misleading, since the
self-doped conducting polymers sounds like the polymers that have been already doped intrinsically without the need of any doping process. However, the self-doped conducting polymers need the doping process to exhibit the electric conductivity. Because the ionic group of the polymer cannot contribute to the electric conductivity, the doping process is still
required to generate solitons, polarons and/or bipolarons. So, for these reasons, Heinze et al.
suggested the term self-ionized conjugated polymers.39Figure 1.3.3 shows the p-doping process (oxidation) for the conducting polymers bearing an anionic group (A-) with a counter cation (usually a small metal cation, M+). When an electron transfer takes place through the conducting polymer backbone, the anionic groups act as the counter ion to the positive charges in ICP, allowing the system of the doped polymer to maintain the overall electrical neutrality. Furthermore, the cation has a higher mobility than the anion in an aqueous system, thereby increasing the rate of the redox process in the self-doped conducting polymer compared to the classical p-type ICPs involving the anion compensation.
X
X X
An(H2C)
n
M
oxidation
reduction X
X X
(CH2)nA
n
+ M
X = N-H or S M = H+, Na+, K+, Li+, etc.
A = ionizable group: SO3-, PO2(OH)-, COO-, etc.
Figure 1.3.3 Charging and discharging processes occurring in a self-doped (ionized) conducting polymer during p-doping39
1.3.3 Various Design of Self-doped ICPs
Self-doped conducting polymers have been prepared by the polymerization of ionic monomers or the post-synthesis of the ICP macromolecular acids such as those bearing sulfonic acids, carboxylic acids, phosphonic acids, and boronic acids. In general, there are two kinds of self-doped conducting polymers. One of them is conducting polymers bearing ionic functional groups, linked covalently to the aromatic rings of the polymer backbones. The other one is those having ionic groups bound to spacer-type heteroatoms in the conjugated backbones. Some examples of polypyrrole, polythiophene and polyaniline derivatives are shown in Figure 1.3.4, Figure 1.3.5 and Figure 1.3.6, respectively.
One of the most important properties of the self-doped conducting polymers is solubility74, and indeed the insoluble problem of ICPs is directly related with their low processability. This is believed to be due to the crystalline polymer structures and the rigidity of the main chains. Apparently, the delocalization of the -system has a tendency to form the
coplanar structure, which makes the polymers inflexible and insoluble. Fortunately, this problem could be overcome by structural modifications. Actually, the ionic groups of the self-doped conducting polymers disrupt the packing forces among the polymer backbones and increase the polymer-solvent interactions.
NH
(CH2)n SO3Na
n N
H
(CH2)n COOH
n N
H
n
SO3H
NH
n
COOH
N ONa O
n
N
SO3-
m
HN
n
(Y+m)+
(A-)Y Polymer Bulletin,1 18, 227. 2
Synthetic Matals,69, 511. 4
New J.Chem,15, 233.
3
Polymer Communication,32, 412.
5
Solid State Ionics,169, 51. 6
Journal of Electroanalytical Chemistry,250, 355.
Figure 1.3.4Various self-doped polypyrrole derivative
S (CH2)n
SO3M
n S
(CH2)n COOH
n S n
O
S n
JACS,109, 1858.1 2
Synthetic Matals,99, 53. 4
Synthetic Metals,142, 251.
3 JACS,120, 5274.
SO3H
O O
O (CH2)4
SO3Na
S S
(H2C)4SO3-
N+
n
S
S n
N+R(CH3)2 CF3SO3-
S S n
(CH2)6N+(CH3)3ClO4- H3C(H2C)5
Polymer Preprints,6 39, 137.
Chem. Mat.,5 9, 2940. 7
Synthetic Metals,119, 153.
Figure 1.3.5 Various self-doped polythiophene derivatives.
HN SO3H
n N n
N n
HN COOH
n SO3H
HN
CH2PO(OH)2
n
HN B(OH)2
n
HN H
N SO3H
0.5 0.5
HN N
0.5 0.5
(CH2)n SO3H
(CH2)n SO3H
HN H
0.5 N 0.5
COOH
HN N
0.5 0.5
HO3S SO3H
HN N
0.5 0.5
SO3H Synthetic Metals, 31,2 369.
Chem.Comm1990,180.
Polymer,133, 4410. 3
Polymer,34, 158. 4
Macromolecules,25, 6029.
Chem.Comm,51995, 1327. 6
JACS,126, 52. 7
JACS,113, 2665.
Macromolecules,24, 4441.
Macromolecules,8 27, 3625. 9
Macromolecules,25, 6029.
JJAP part 2,1033, L357. 11
Macromolecules,27, 3625.
Figure 1.3.6Various self-doped polyaniline derivatives.
Polyelectrolyte and ICPs
The polyelectrolytes or polymeric acids (polyanions) that have been used as dopants to prepare water-soluble conducting polymers include poly(ethanesulfonic acid), poly(acrylic acid), poly(styrenesulfonic acid), and poly(amic acid). Instead of acid, these polymeric acids can be incorporated into the conducting polymers.
Polyanilines with polyelectrolytes have been developed. In the presence of polyelectrolytes such as poly(4-styrenesulfonate) and poly(vinylphosphonic acid) under aqueous buffer conditions (pH 4.3), aniline monomers were polymerized. During the polymerization, polyelectrolytes acted as a template. Due to the electrostatic and hydrophobic interactions between the polyelectrolyte and the aniline derivatives, a complex formation of the macromolecular polyelectrolyte template proceeded along the polyaniline chain.71 Consequently, the resulting polyaniline derivatives were soluble in water.
According to the similar process, Bae et al. reported water-soluble polypyrrole graft copolymers.72-73 The most different point as compared with the above-mentioned macromolecular complex was the presence of the covalent bond with the poly(styrenesulfonic acid) chain as shown in Figure 1.3.7.
N
NH2 HN
NH
HN NH2
HN NH
SO3H SO3H SO3 SO3 SO3H SO3H SO3
Figure 1.3.7The structure of self-doped polypyrrole graft copolymer73
Lukkari et al. firstly reported on the all thiophene-type polyelectrolyte, consisted of a polyanion, poly(3-(3-thienyloxy)propanesulfonate) (P3TOPS), and a polycation, poly(3-(3-thienyloxy)propyl triethyl ammonium) (P3TOPA), as shown in Figure 1.3.8. They fabricated the multilayers, and demonstrated the linear charge density of the conducting polymer.75
S n SO3-
P3TOPS
S n
N+
P3TOPS
Figure 1.3.8Structures of P3TOPS and P3TOPA75
Cao et al. showed the photovoltaic properties of a multilayer film based on highly sulfonated polyaniline and diazoresin.76Chanet al.fabricated multilayer photovoltaic devices based on ruthenium-containing poly(p-phenylenevinylene) and sulfonated polyaniline.77
1.3.4 Self-doped ICPs as Rechargeable Battery
Most of the common conducting polymers exchange anions during the redox processes, i.e., the charging and discharging processes, when they are applied as cathodes vs. the Li metal anode. However, self-doped ICPs exclude the participation of anions in the charging
discharging process. Instead, the charge compensation occurs with involving the migration of a lithium cation, which helps to minimize the electrolyte volume, resulting in downsizing the overall battery and maximizing the energy density. Note that the anion migration during the charging/discharging process brings about a change in the salt concentration; consequently, the electrolyte must be provided in amounts larger than the equivalent amount required for the charge compensation to maintain the ionic conductivity of the electrolyte layer, and hence excess electrolyte is unavoidable. The copolymer ofo-aminobenzenesulfonic acid and aniline was reported to exhibit a specific charge of ca. 47 Ah kg 1 in aqueous solutions and an even larger value over 80 Ah kg 1was expected in nonaqueous solutions.80
Furthermore, sulfonated polyanilines are typical cathodic materials in secondary battery applications. Steric protection by the sulfonic group helps to reduce the degradation of the quinoid structure during oxidation, resulting in the improved cyclic performance.81-82
1.4 Facilitated Transport Phenomena
1.4.1 Introduction
The carrier-mediated mass transport phenomena are observed in a living body, where oxygen reacts with hemoglobin in lung to make oxygen-heme complexes, followed by flowing through blood vessels, and is released in organs. Subsequently de-complexed hemoglobins return back to the lung to make oxygen-heme complex again. Here transport properties of oxygen are mostly determined by the flowability of the complexes through blood vessels, where a liquid blood or solvent is necessary to carry carrier-solute complexes.
Here heme structure in the hemoglobin reacts selectively with oxygen reversibly, thus called an oxygen carrier.83
Facilitated transport is defined as a carrier-mediated transport phenomenon by means of coupled diffusion and reversible reaction. The carrier forms a reversible complex with a specific component relative to others, thereby increasing the transport rate of the component relative to others. Therefore, this concept is a very promising to develop a permselective mass transport medium, for example, air separation with metallo-porphyrins or metal-Schiff's bases as shown in Figure 1.4.1,84acid gas separation with amine carriers and olefin separation with silver ion85 or silver metal.86 Furthermore, it could be applied to solid electrolyte in a Li-ion battery.
Figure 1.4.1Reversible oxygen coordination to iron (II) porphyrin.84
1.4.2 Developed mathematical models
Mathematical models have been developed to analyze the facilitation phenomena in a fixed site carrier membrane. Tsuchida et al. suggested that dual sorption model has been commonly employed, because it is conceptually analogous to the mass transport in a facilitated transport membrane with fixed site carriers and it has a mathematical simplicity.87 However, it does not predict facilitated transport without direct diffusion between carriers.
Noble introduced a concept of "the effective diffusion coefficient" between fixed site
carriers.88 It failed to predict the case of high flux or high pressure, since the starting assumption is the excess carrier sites. Cussler et al. suggested a concept of limited mobility of chained carriers.89 Their assumption is as follows; 1) there is no uncomplexed solute in the membrane, and 2) the reaction is fast between a carrier and solute at the surface of the membrane. In parell to "the effective diffusion coefficient", it also failed to explain the condition at low flux or pressure.
A reversible complex reaction continuously occurs at carrier/solute in facilitated transport membranes. Owing to the reversible reaction, the momentary concentration of the solute could be fluctuated at the carrier site. Nevertheless, macroscopic time average concentration can be represented as a line in Figure 1.4.2.90 The concentration fluctuation leads to the increase in the chemical potential of the solute according to Cahns theory.91
Figure 1.4.2 Fluctuated concentration profile in solid state facilitated transport membrane at steady state. (where P0is permeability of the membrane matrix without carrier. pdis pressure fluctuation due to the reversible reaction.)90
The increased chemical potential leads to a higher driving force for mass transfer through the membrane. Taking a fluctuated concentration concept, Kang et al. proposed a concentration fluctuation model to explain the facilitated transport behavior in the solid state.
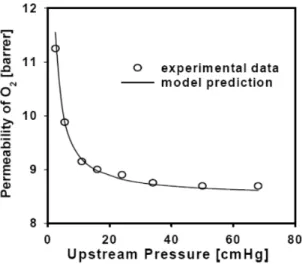
The concentration fluctuation model was examined against the experimental data on facilitated oxygen transport through PBMA containing cobalt porphyrins. The model predicts the experimental results exceptionally well even at high pressure range where the effective diffusion coefficient model fails to predict. This suggests the validity of the concentration fluctuation model.
Figure 1.4.3 The comparison between the concentration fluctuation model predictions and experimental data for PBMA membrane containing cobalt porphyrin90
1.4.3 Olefin separation membrane
Facilitated olefin transport membranes have been considered as an alternative to the distillation for olefin/paraffin separation. In the case of propylene transport, there are many reports about silver polymer electrolyte membranes. The solid silver-polymer complex membranes have been developed by dissolving silver salts of low lattice energy in a polymer matrix via coordination bond, and they are successfully used as facilitated olefin transport membranes in the solid state. Silver polymer complex membranes containing AgBF4 or AgCF3SO3 dissolved either in poly(2-ethyl-2-oxazoline) (POZ), in poly(N-vinylpyrrolidone) (PVP) or in poly(ethylene oxide) (PEO) exhibit facilitated olefin transport, resulting in high olefin separation performance from its mixture with paraffin. For example, a thin composite membrane comprising AgBF4dissolved in POZ or PVP is approximately 15,000 times more permeable to propylene than propane in pure gas permeation because of the reversible complexation between the silver ion and propylene,i.e., the facilitated propylene transport.85
The silver nanoparticles (AgNPs) which have partially positive charge on its surface have been demonstrated as a carrier in facilitated propylene transport. For example, poly(ethylene-co-propylene) (EPR)/AgNPs/p-benzoquinone composite membranes demonstrated a propylene/propane selectivity of 11 and a total mixed gas permeance of 0.5 GPU (1 GPU = 1 × 106 cm3 (STP)/(cm2 s cmHg)). In these cases, an electron acceptor, p-benzoquinone (p-BQ) caused the surface of silver nanoparticles to be partially positively polarized.93-94Here, surface-activated silver was demonstrated as a carrier center.
References
1. M. Armand, J.-M. Tarascon,Nature,2008,451, 652.
2. F. K. Lutgens, E. J. Tarbuck,Essentials of Geology 7th ed, Prentice Hall, New York, 2000.
3. A. G. MacDiarmid, L. S. Yang, W. S. Huang, B. D. Humphrey,Synth. Metals,1987, 18, 393.
4. H. Nishide, K. Oyaizu,Science,2008,319, 737.
5. K. Oyaizu, H. Nishide,Adv. Mater,2009,21, 2339.
6. T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu, H. Nishide,Adv. Mater,2009,21, 1627.
7. I. S. Chae, M. Koyano, K. Oyaizu, H. Nishide,J. Mater. Chem. A,2013,1, 1326.
8. J. S. Miller,Adv. Mater.1993,5, 671.
9. K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro, E. Hasegawa, Chem. Phys. Lett.2002,359, 351.
10. O. Piloty and B. G. Schwerin,Berichte der Deutschen Chemischen Gesellschaft,1901, 34, 1870.
11. A. N. Holden, W. A. Yager and F. R. Merity,J. Chem. Phys.,1951,19, 1319.
12. (a) H. Wieland and M. Offenb¨acher,Berichte der Deutschen Chemischen Gesellschaft, 1914,47, 2111; (b) H. Wieland and K. Roth,Berichte der Deutschen Chemischen Gesellschaft,1920,53, 210.
13. K. H. Meyer and W. Reppe,Berichte der Deutschen Chemischen Gesellschaft,1921,54, 327.
14. O. L. Lebedev, M. I. Khidekel and G. A. Razuvaev,Doklady Akademii Nauk SSSR, 1961,140,1327.
15. (a) E. G. Rozantsev, V. D. Sholle,Synthesis,1971, 190; (b) E. G. Rozantsev ,V. D.
Sholle,Synthesis,1971, 410; (c) E. G. Rozantsev, V. D. Sholle,Synthesis,1984, 895.
16. E. Breuer, H. G. Aurich and A. Nielsen,Nitrones, Nitronates, and Nitroxides, John Wiley and Sons Ltd, Chichester,1989.
17. H. B. Hass and E. F. Riley,Chem. Rev.,1943,32, 373.
18. L. R. Mahoney, G. D. Mendenhall and K. U. Ingold,J. Am. Chem. Soc.,1973,95, 8610.
19. T. Suga, Y. J. Pu, K. Oyaizu, H. Nishide,Bulletin of the Chemical Society of Japan 2004,77, 2203.
20. A. Capiomont, B. Chion, J. Lajzerowicz,Acta Cryst. Sect. B,1971,27, 322.
21. Y. Yonekuta, K. Oyaizu, H. Nishide,Chem. Lett.2007,36, 866.
22. K. Oyaizu, Y. Ando, H. Konishi, H. Nishide,J. Am. Chem. Soc.2008,130, 14459.
23. K. Koshika, N. Chikushi, N. Sano, K. Oyaizu, H. NishideGreen. Chem.,2010,12, 1573.
24. T. Suga , Y. J. Pu , S. Kasatori , H. Nishide ,Macromolecules2007,40, 3167.
25. M. Suguro, S. Iwasa, Y. Kusachi, Y. Morioka, K. Nakahara,Macromol. Rapid Commun.2007,28, 1929.
26. M. Suguro, S. Iwasa, K. Nakahara,Macromol. Rapid Commun.2008,29, 1635.
27. K. Oyaizu, T. Kawamoto, T. Suga, H. Nishide,Macromolecules2010,43, 10382.
28. J. Qu, R. Morita, M. Satoh, J. Wada, F. Terakura, K. Mizoguchi, N. Ogata, T. Masuda, Chem. Eur. J.2008,14, 3250.
29. K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh, E. J. Cairns,J. Power Sources 2007,163, 1110.
30. T. Suga , Y. J. Pu , S. Kasatori , H. Nishide ,Macromolecules2007,40, 3167.
31. (a) P. Nesvadba , L. Bugnon , P. Maire , P. Novak ,Chem. Mater.2010,22, 783. (b) T.
Suga, H. Ohshiro, S. Sugita, K. Oyaizu, H. Nishide,Adv. Mater.2009,21, 1627. (c) H.
Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara, M. Satoh,Electrochim. Acta2004, 827. (d) T. Suga, H. Konishi, H. Nishide,Chem. Commun.2007, 1730.
32. A. Pron, P. Rannou,Progress in Polymer Science2002,27, 135.
33. T. Shimidzu, A. Ohtani, T. Yyoda, K. Honda,J. Electroanal. Chem.1987,224, 123.
34. M. Lien, W. H. Smyrl, M. Morita,J. Electroanal. Chem.1991,309, 333.
35. K. Naoi, M. Lien, W. H. Smyrl,J. Electrochem. Soc.1991,138, 440.
36. G. Bidan, B. Ehui, M. Lapkowski,J. Phys. D: Appl. Phys.1988,21, 1043.
37. M. S. Freund, B. Deore,Self-Doped Conducting Polymers; Wiley (Chichester), 2007.
38. A. O. Patil, Y. Ikenoue, N. Basescu, N. Colaneri, J. Chen, F. Wudl, A. J. Heeger, Synthetic Metals1987,20, 151.
39. Jurgen Heinze, B.A. Frontana-Uribe, S. Ludwigs,Chem. Rev.2010,110,4724.
40. C. Dearmitt, S. P. Armes, J. Winter, F. A. Uribe, S. Gottesfeld, C. Mombourquette, Polymer1993,34,158.
41. M. T. Nguyen, P. Kasai, J. L. Miller, A. F. Diaz,Macromolecules,1994,27, 3625.
42. H. S. O. Chan, S. C. Ng, W. S. Sim, K. L. Tan, B. T. G. Tan,Macromolecules1992,25, 6029.
43. B. Deore, M. S. Freund,Analyst2003,128, 803.
44. B. A. Deore, I. Yu, M. S. Freund,J. Am. Chem. Soc.2004,126, 52.
45. J. Yue, Z. H. Wang, K. R. Cromack, A. J. Epstein, A. G. MacDiarmid,J. Am. Chem.
Soc.1991,113, 2665.
46. J. Yue, A. J. Epstein,Macromolecules1991,24, 4441.
47. T. Kawai, H. Mizobuchi, N. Yamasaki, H. Araki, K. Yoshino,Japanese Journal of Applied Physics Part 2 – Letters1994,33, L357.
48. R. S. Wang, L. M. Wang, Z. M. Su, Y. J. Fu,Synthetic Metals1995,69, 511.
49. E. T. Kang, K. G. Neoh, Y. L. Woo, K. L. Tan,Polymer Communications1991,32,412.
50. D. Delabouglise, F. Garnier,New Journal of Chemistry1991,15, 233.
51. M. D. Ingram, H. Staesche, K. S. Ryder,Solid State Ionics2004,169, 51.
52. D. J. Liaw, B. Y. Liaw, J. P. Gong, Y. Osada,Synthetic Metals1999,99, 53.
53. M. Chayer, K. Faid, M. Leclerc,Chemistry of Materials1997,9, 2902.
54. K. Faid, M. Leclerc,J. Am. Chem. Soc.1998,120, 5274.
55. F. Tran-Van, M. Carrier, C. Chevrot,Synthetic Metals2004,142, 251.
56. G. Zotti, S. Zecchin, G. Schiavon, A. Berlin,G. Pagani, A. Canavesi,Chemistry of Materials1997,9, 2940.
57. A. A. Moxey, D. C. Loveday, I. D. Brotherson, J. P. Ferraris,Polymer Preprints (American Chemical Society, Division of Polymer Chemistry)1998,39, 137.
58. A. Berlin, G. Schiavon, S. Zecchin, G. Zotti,Synthetic Metals2001,119, 153.
59. Y. Cao, P. Smith, A. J. Heeger,Synthetic Metals1993,57, 3514.
60. M. Lapkowski,Synthetic Metals1993,55,1558.
61. M. Angelopoulos, N. Patel, R. Saraf,Synthetic Metals1993,55, 1552.
62. S. K. Sahoo, R. Nagarajan, S. Roy, L. A. Samuelson, J. Kumar, A. L. Cholli, Macromolecules2004,37, 4130.
63. W. J. Bae, K. H. Kim, Y. H. Park, W. H. Jo,Chem. Comm.2003, 2768.
64. W. J. Bae, K. H. Kim, W. H. Jo, Y. H. Park,Macromolecules2005,38, 1044.
65. X. L. Wei, Y. Z. Wang, S. M. Long, C. Bobeczko, A. J. Epstein,J. Am. Chem. Soc.
1996,118, 2545.
66. H. K. Lin, S. A. Chen,Macromolecules2000,33, 8117.
67. K. Takahashi, K. Nakamura, T. Yamaguchi, T. Komura, S. Ito, R. Aizawa, K. Murata, Synthetic Metals2002,128, 27.
68. M. Leclerc, K. Faid,Adv. Mater.1997,9, 1087.
69. M. T. Nguyen, M. Leclere, A. F. Diaz,Trends in Polymer Science1995,3, 186.
70. N. Zhang, R. Wu, Q. Li, K. Pakbaz, C. O. Yoon, F. Wudl,Chem. Mater.1993,5, 1598.
71. S. K. Sahoo, R. Nagarajan, S. Roy, L. A. Samuelson, J. Kumar, A. L. Cholli, Macromolecules2004,37, 4130.
72. W. J. Bae, K. H. Kim, Y. H. Park, W. H. Jo,Chem. Comm.2003, 2768.
73. W. J. Bae, K. H. Kim, W. H. Jo, Y. H. Park,Macromolecules2005,38, 1044.
74. M. Ballauff,Angew. Chemie,1989,28, 253.
75. J. Lukkari, A. Viinikanoja, J. Paukkunen, M. Salomaki, M. Janhonen, T. Aaritalo, J.
Kankare,Chem. Comm.2000, 571.
76. T. B. Cao, L. H. Wei, S. M. Yang, M. F. Zhang, C. H. Huang, W. X. Cao,Langmuir 2002,18, 750.
77. K. Y. K. Man, H. L. Wong, W. K. Chan, C. Y. Kwong, A. B. Djurisic,Chem. Mater.
2004,16, 365.
78. Y. Ikenoue, H. Tomozawa, Y. Saida, M. Kira, H. Yashima,Synthetic Metals1991,40, 333.
79. G. Sonmez, I. Schwendeman, P. Schottland, K. W. Zong, J. R. Reynolds, Macromolecules,2003,36, 639.
80. C. Barbero, R. Koetz,Adv. Mater.1994,6, 577.
81. C. Barbero, M. C. Miras, B. Schnyder, O. Haas, R. Kotz,Journal of Materials Chemistry1994,4, 1775.
82. M. S. Rahmanifar, M. F. Mousavi, M. Shamsipur,Journal of Power Sources2002,110, 229.
83. Y. S. Kang, Solid State Facilitated Transport Membranes
84. E. Tsuchida, H. Nishide, M. Ohyanagi, H. Kawakami,Macromolecules1987,20, 1907.
85. Y. Yoon, J. Won, Y. S. Kang,Macromolecules2000,33, 3185.
86. I. S. Chae, S. W. Kang, J. Y. Park, J. H. Lee, J. Won, and Y. S. Kang,Angew. Chem.
Int. Ed.2011,50, 2982.
87. H. Nishide, M. Ohyanagi, O. Okada, E. Tsuchida,Macromolecules1987,20, 417.
88. R. D. Noble,J. Membr. Sci.1990,50, 207.
89. E. L. Cussler, R. Aris, A. Bhown,J. Membr. Sci.1989,43, 149.
90. Y. S. Kang, J. M. Hong, U. Y. Kim, J. Jang,J. Membr. Sci.1996,109, 149.
91. J. W. Cahn,J. Chem. Phys.1965,42, 93.
92. M. Ohyangai, H. Nishide, K. Suenaga, E. Tsuchida,Macromolecules1988,21,1590.
93. Y. S. Kang, S. W. Kang, H. Kim, J. H. Kim, J. Won, C. K. Kim, K. Char,Adv. Mater.
2007,19, 475.
94. S. W. Kang, K. Char, Y. S. Kang,Chem. Mater.2008,20, 1308.
Self-doping Inspired Zwitterionic Pendant Design of Radical
Polymers toward a Rocking-chair-type Organic Cathode-active Material
2.1 Introduction
2.2 Preparation of Poly(TEMPO-methacrylate-stat-vinyl sulfonic acid) 2.3 The electrochemical properties involving cation migration
2.4 Experimental Section References