111
hp170010 「京」以外 HPCI 産業利用(実証利用) HPCI other than K Industrial Use
鉛と硫酸水溶液界面の第一原理分子動力学計算
Ab initio molecular dynamics simulations of interface between lead and sulfuric
acid aqueous solution
窪田 善之 Yoshiyuki Kubota
関西電力(株) 技術研究所
Research and Development Center, The Kansai Electric Power Company, Inc.
要旨 不可逆の硫酸化は,鉛蓄電池の負極のよく知られた劣化形態である。水和された硫酸イオンと 鉛表面の間の反応性は,分子の観点から硫酸化を理解するために第一原理分子動力学計算を使っ て調べられた。分子動力学計算は,300 K で 50 ps の分子動力学時間の間,水和された硫酸イオン と鉛表面の間に約 0.4 nm の隙間をともなって無反応を示した。厚板模型の鉛原子は,相対的に大 きな位置の揺動を示した。 キーワード:第一原理分子動力学計算,鉛蓄電池,鉛表面,硫酸水溶液 Abstract
The irreversible sulfation is a well-known failure mode for the negative electrode of lead acid battery. The reactivity between the hydrated sulfate and lead surfaces was investigated using ab initio molecular dynamics (AIMD) simulations to understand the sulfation in the molecular view. The molecular dynamics simulations showed no reaction with a gap of ca. 0.4 nm between the hydrated sulfate and the lead surfaces during the molecular dynamics time of 50 ps at 300 K. The lead atoms of the slab model indicated a relatively large positional fluctuation.
Keywords:Ab initio molecular dynamics, Lead-acid battery, Lead surfaces, Sulfuric acid aqueous solution
© 2019 Research Organization for Information Science and Technology All rights reserved. Received: 29 March 2019
Accepted: 16 December 2019 Available online: 26 December 2019
112 1. 研究の背景と目的 鉛蓄電池 (PbAB) は自動車のバッテリー,電力・通信分野の無停電電源装置などに数多く使用 されている二次電池である[1]。PbAB の正極材料には二酸化鉛 (PbO2) が,負極材料には鉛 (Pb) が,電解液には希硫酸水溶液 (H2SO4(aq))が使用される。PbAB の電気化学反応式は,以下のよう に表される。 (正極) PbO2 + SO42- + 4H3O+ + 2e- ⇄ PbSO4 + 6H2O (1) (負極) Pb + SO42- ⇄ PbSO4 + 2e- (2) 放電時には右矢印の,充電時には左矢印の反応が起きる。放電生成物は硫酸鉛 (PbSO4) である。 PbAB のおもな劣化要因は負極のサルフェーション,正極の軟化などが知られている[2]。負極の サルフェーションとは,放電時に生成する硬い PbSO4の形成によって,充電時に Pb に還元され ない不具合のことである。近年,パルス電圧の印加[3]や逆充電[4]により負極のサルフェーション が生成した PbAB を再生させる方法が提案されている。しかしながら,PbAB の再生の機構はお ろか,サルフェーションの生成機構すらわかっていないため,このような再生技術の定量的な評 価が進んでいない。そこで,本研究ではサルフェーションの生成機構を調べる第 1 ステップとし て,PbAB の負極活物質である Pb と電解液である H2SO4(aq)の界面の状態を原子・分子レベルの 観点から調べる。 2. 計算モデル
すべての計算は Viena Ab initio Simulation Package (VASP)の version 5.4.1[4-8]を使って実行され た。電子状態は密度汎関数理論 (DFT) に基づいて計算された。原子のイオンコアを表現するた めに Project augmented wave ポテンシャル[9,10]が使われた。交換相関項は Perdew らの一般化勾 配近似 (PBE) [11]を,van der Waals 相互作用は DFT-D3[12,13]を用いた。平面波基底のカットオ
フエネルギーは 400 eV とし,Pb 表面と H2SO4(aq)の界面の模型の Brillouin ゾーンは𝑀𝑀1× 𝑀𝑀2 k
点によってサンプリングされた。ここで𝑀𝑀1 と 𝑀𝑀2 は Pb 表面に垂直な方向の格子ベクトル𝒂𝒂1と
𝒂𝒂2に対して (𝑀𝑀𝑖𝑖|𝒂𝒂𝑖𝑖|)−1≅ 0.03 Å−1となるように設定された。イオンの時間発展は Born–
Oppenheimer 分子動力学計算によって,カノニカル NVT アンサンブルで実行された。ab initio molecular dynamics (AIMD) の時間ステップは 1 fs と設定された。系の温度は Nosé–Hoover 熱浴 [14,15]により制御され,標的温度は 300 K とした。平衡化計算は全ポテンシャルエネルギーの平 均値が一定値に収束するまで実行された。 最初に Pb の基本物性を評価するためにバルク Pb 結晶の電子状態計算が実行された。バルク Pb 結晶の物性は Birch–Murnaghan 状態方程式[16]を使って計算した。得られた Pb 結晶の平衡格 子定数を用いて,真空層が 2 nm である 3 種類の表面(100),(110),(111)のスラブ模型を構築し, 表面エネルギーの観点から Pb スラブの積層数を評価した。バルク Pb の基本物性と Pb の表面エ ネルギーについては,最外殻の 6s6p 軌道の電子を考慮した計算と 5d 軌道まで含めた計算の両方
113 を実行した。Pb 表面と H2SO4(aq)の界面の模型では,表面に平行な方向のスーパーセルの長さが 約 1 nm になるように設定した。H2SO4(aq)層は無限希釈を想定し,硫酸イオン (SO42–) 1 個とヒド ロニウムイオン (H3O+) 2 個が水溶液中に置かれ,SO42–イオンは Pb 表面の直上に位置するように 配置した。H2SO4(aq)の密度の実験値[17]を使って,水溶液の厚さが約 2 nm になるように水分子 の数が決められた。最小化計算は,Pb 原子の位置を全て固定して実行した。最小化計算後の温度 上昇計算と平衡化計算はすべての原子を緩和して実行した。平衡化計算は,全ポテンシャルエネ ルギーの平均値が一定値を中心にゆらぐまで実行した (約 3~4 ps の AIMD 計算) 。使用された スーパーセルの大きさとスーパーセル内に含まれる Pb 原子,硫酸分子,H2O 分子の数は Table 1 に示されている。 3. 並列計算の方法と効果(性能) AIMD 計算の効率的な並列数を評価するために,東京大学のスーパーコンピュータ Reedbush-U の並列ノード数とバンドあたりに割り振るコア数を決定する VASP の変数 NPAR を変化させる ことにより AIMD 計算を 2 ステップ実行した。AIMD 計算にかかる計算時間のみを評価するため に,波動関数ファイル WAVECAR はあらかじめ作成し,最初の電子状態計算ではそれを読み込む 設定 (ISTART = 1) として,電荷ファイルの出力なし (LCHARG = False) と WAVECAR の出力な し(LWAVE = False)の設定として,Elapsed Time を評価した。Table 2 は本計算系についての Reedbush-U での AIMD 計算のパフォーマンスを示す。本研究で考慮した全ての表面と全てのノ ードで,NPAR = 18 が最も経過時間が短かった。ノード時間積と経過時間の観点から,全ての表 面で並列ノード数 = 5 と NPAR = 18 が採用された。 4. 研究成果 バルク Pb 結晶の物性値の計算結果と実験値の比較が Table 3 に示されている。我々の PBE–D3 の格子定数と体積弾性率の予測値は,以前の PBE の計算結果[18,19]より実験値[20,21]に近かっ た。一方,PBE–D3 の凝集エネルギーは,PBE のそれ[18]に比べて実験結果[21]を過大評価した。
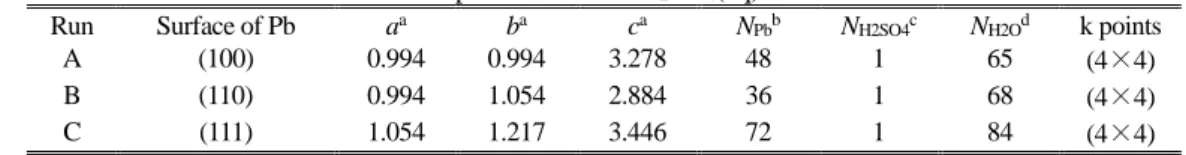
Table 1 Supercells used in H2SO4(aq) / Pb models.
Run Surface of Pb aa ba ca N
Pbb NH2SO4c NH2Od k points
A (100) 0.994 0.994 3.278 48 1 65 (4×4) B (110) 0.994 1.054 2.884 36 1 68 (4×4) C (111) 1.054 1.217 3.446 72 1 84 (4×4)
aParameters of supercell (nm). bNumber of Pb atoms in supercell. cNumber of H
2SO4 molecule in supercell. dNumber of H
2O molecules in supercell.
Table 2 Performances obtained using the supercomputer Reedbush-U in the University of Tokyo.
Node 4 5 6 NPAR 12 18 36 12 18 36 12 18 36 (100) 84.3 77.5 85.4 76.4 68.7 76.5 71.7 64.5 71.8 ta (110) 65.7 60.9 63.5 60.4 55.1 58.3 58.9 52.5 57.3 (111) 178.4 153.2 171.5 148.2 133.8 150.6 136.3 124.7 139.1 aElapsed time (s).
114 5d 軌道の電子を考慮すると,5d 軌道の電子を考慮しない場合に比べて格子定数,体積弾性率, 凝集エネルギーの予測値が実験結果に近づくことがわかった。Pb の低指数表面の表面エネルギ ーは過去の DFT 計算により繰り返し調べられた。Table 4 は過去の DFT 計算と実験の表面エネル ギーの比較を示す。本研究で実施された Pb の表面エネルギーの収束性については,付録 A の Fig. A1 を参照されたい。Table 4 に示されている本研究での表面エネルギーは,6 層積層したスラブ によって得られた値である。DFT 計算によって予測された Pb 表面の安定性の順[18,19,23]は,(111) > (100) > (110)であり,実験結果[24]を再現する。過去の DFT 計算の間の表面エネルギーの比較か ら,s と p 軌道のみを価電子として考慮したノルム保存擬ポテンシャル (NCPP) 法による局所密 度近似 (LDA) [18]が最も実験結果[24]に近く,フルポテンシャル法 (FP KKR) による LDA[23]が 最も実験値[24]から遠いことがわかる。Yu らによって指摘されたように PBE の表面エネルギー は LDA のそれに比べて小さい[18]。本研究で得られた PAW–PBE–D3 の結果と Li らの PAW–PBE の結果[19]を比較すると,分散力は表面エネルギーの増加に寄与することがわかった。価電子へ の 5d 軌道の電子の含有は表面エネルギーをわずかに減少させる。
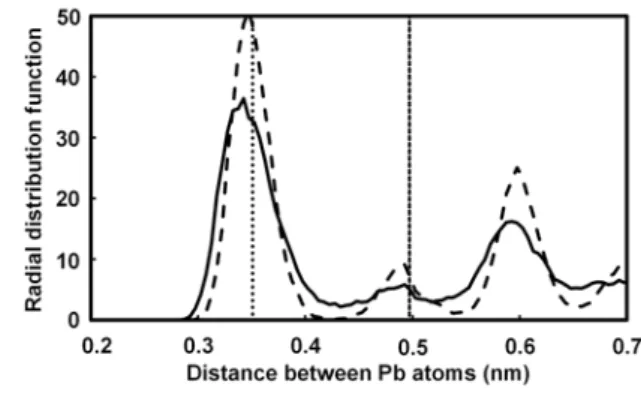
最初に 300 K の温度での Pb の真空層スラブ模型の AIMD 計算を実行した。その結果,表面と バルク内部の Pb 原子がともに大きく移動した。Figure 1 は,Pb(110)面のスラブ模型の Pb 原子間 距離の動径分布関数 (Radial distribution function: RDF) を示す。第 1 近接 (0.351 nm) ,第 2 近接 (0.497 nm) のバルク Pb の原子間距離が点線で表されている。表面と温度の効果が,半値幅が約 0.1 nm の第 1 近接のピークを与えることがわかった。Pb(100)と(111)面の RDF は,Pb(110)面のそ れとほとんど同じであった。
次に上述の真空層に H2SO4(aq)を挿入した模型を構築し,300 K の温度での AIMD 計算を実行
した。Figure 1 に示されるように Pb の真空層スラブ模型の AIMD 計算と比較して,H2SO4(aq)の
影響により Pb 原子が大きく揺らぎ,第 1 近接と第 2 近接のピークの高さが低くなることがわか Table 3 Physical properties of bulk Pb.
a0a B0b B0’c Ecohd PAW–PBE–D3(5d)e 0.494 43 6 3.71 PAW–PBE–D3f 0.497 44 6 3.74 NCPP–LDAg 0.485 53 5 3.83 NCPP–PBEg 0.506 39 5 2.99 PAW–PBEh 0.503 41 – 2.99 Expt. 0.491i 43j 5j 2.03k aLattice parameter (nm).
bBulk modulus (GPa). cPressure derivative of B
0. dCohesive energy (eV/f.u.).
eThis work. The inner 5d orbitals are included
in valence states.
fThis work. The inner 5d orbitals are not included
in valence states. gReference 18. hReference 19. iReference 20 (298 K). jReference 21. kReference 22.
Table 4 Surface energies of Pb (mJ m–2).
Pb(111) Pb(100) Pb(110) PAW–PBE–D3(5d)a 458 492 533 PAW–PBE–D3b 474 511 551 FP KKR–LDAc 600 640 720 NCPP–LDAd 417 479 493 NCPP–PBEd 276 320 333 PAW–PBEe 275 321 337 Expt. (323 K)f 441 468 482 aThis work. The inner 5d orbitals are included
in valence states.
bThis work. The inner 5d orbitals are not included
in valence states.
cReference 23. Experimental lattice parameter was used. dReference 18.
eReference 19. fReference 24.
115
った。さらに H2SO4(aq)をともなった Pb スラブでは,第 1 近接のピーク位置が原子間距離の縮小
方向にシフトした。これは H2SO4(aq)層によって,Pb スラブが圧縮されたためであると考えられ
る。Table 5 は H2SO4(aq)層をともなった Pb(100),(110),(111)面の AIMD 計算によって得られた
SO42–イオンに関係した距離を示す。Pb 表面に垂直な方向 (スーパーセルの z 軸) については,(3) 式に示される距離の平均値,𝑑𝑑𝑍𝑍_aveを計算した。 𝑑𝑑𝑍𝑍_ave= 〈𝑍𝑍S(𝑡𝑡) −𝑁𝑁1 Surf.Surf.� 𝑍𝑍Pb,𝑖𝑖(𝑡𝑡)〉 (3) ここで,𝑍𝑍S(𝑡𝑡)と𝑍𝑍Pb,𝑖𝑖(𝑡𝑡)は,MD 時刻 t での S 原子の Cartesian Z 座標の値と i 番目の Pb 原子の Cartesian Z 座標の値である。𝑁𝑁Surf.はスーパーセル内の Pb 表面第 1 層の Pb 原子の数である。Pb 表面内での SO42–イオンの移動距離については,𝑑𝑑𝑍𝑍_aveの値から考えて Pb スラブの影響がほとん どないと考えられるため,(4)式に示される距離の最大値,𝑑𝑑𝑋𝑋𝑋𝑋_maxを計算した。 𝑑𝑑𝑋𝑋𝑋𝑋_max= max𝑡𝑡 ��𝑋𝑋S(𝑡𝑡) − 𝑋𝑋S(0)�2+ �𝑌𝑌S(𝑡𝑡) − 𝑌𝑌S(0)�2 (4) ここで,𝑋𝑋S(𝑡𝑡)と𝑌𝑌S(𝑡𝑡)は MD 時刻 t での S 原子の Cartesian X 座標と Y 座標の値である。50 ps の MD 時間では,SO42–イオンは Pb 表面上 0.37~0.49 nm の位置で維持され,Pb 表面上に近づくこ とはなかった。加えて,SO42–イオンはスーパーセルの xy 平面内の初期位置からほとんど移動す ることはなかった。水分子は SO42–イオンと比べて,一時的に Pb 表面に接近することはあったが, 安定的な Pb-O 結合を形成することはなかっ た。Pb 表面上の H2SO4(aq)中の化学種の振る 舞いは,最近,AIMD 計算を使って実行され た PbAB の正極材料である PbO2表面上の H2SO4(aq)中の化学種の振る舞い (SO42–イオ ンは,PbO2表面上に容易に吸着し,PbO2表面 は H2SO4(aq)の水分子,水酸化物イオン,水素 イオンによって終端される) [25]と全く異なる。 これらの結果から,両電極材料に対する H2SO4(aq)中の化学種の反応性の違いは,電極 材料表面の電荷の局在の違いによるものと考 えられる。Pb 表面と H2SO4(aq)の化学種との 反応性については,これまで理論的および実 験的な報告がなく,本研究で初めて明らかに なった。 ここで,AIMD 計算が Pb 表面と H2SO4(aq) の間で無反応を観測したことについて,考え られる要因を列挙する。1 つ目は短すぎる MD 継続時間。本研究では 50 ps という MD 継続 Table 5 Distances relevant to SO42–anion computed using
AIMD simulation at 300 K.
Pb(111) Pb(100) Pb(110) 𝑑𝑑𝑍𝑍_avea 0.369±0.022 0.389±0.026 0.486±0.059
𝑑𝑑𝑋𝑋𝑋𝑋_maxb 0.252 0.168 0.088
aAverage distance between S atom and the Pb surface
expressed by Eq. (3) (nm).
bMaximum distance from initial position for S atom
expressed by Eq. (4) (nm).
Fig. 1 Radial distribution functions as a function of Pb
atomic distance at 300 K for Pb(110) slab with H2SO4(aq) (solid curve) and vacuum (dashed curve).
Vertical dotted lines indicate the nearest (0.351 nm) and the second nearest (0.497 nm) neighboring distances.
116
時間で AIMD 計算が実行された。AIMD 計算の延長により,反応が起きるかもしれない。2 つ目 は電圧を考慮していないこと。電場を考慮することにより電極近傍の電解液は,電気二重層を構 成し,反応性が高められるかもしれない。3 つ目は放電を考慮していないこと。通常,充電状態
にある PbAB では,放電によって負極 Pb が PbSO4に化学変化する。PbAB の自然放電が起きない
と仮定するならば,放電過程を考慮しなければ,負極 Pb は化学変化しない。すなわち,本研究 の AIMD 結果は,自然放電を考えない充電状態の PbAB の挙動と矛盾するものではない。したが って,私は 3 つ目の PbAB の放電を考慮していないことが,無反応の一番の要因であると推察す る。 5. まとめと今後の課題 PbAB の負極活物質である Pb と電解液である硫酸水溶液の界面を模擬した模型を用いて, AIMD 計算を実行した。本研究で調べた 3 種類のいずれの Pb 表面でも,50 ps の MD 時間では, H2SO4(aq)のイオンあるいは分子が Pb 原子と反応を起こさなかった。真空層スラブ模型を用いた AIMD 計算は,表面の Pb 原子とバルク内部の Pb 原子がともに大きく揺らぐことを観察した。真 空層が H2SO4(aq)で置き換えられた模型では,真空層スラブ模型に比べて,さらに大きく Pb 原子 が揺らぐことがわかった。 PbAB の負極 Pb と H2SO4(aq)が反応を起こすためには,放電を模擬する,すなわち Pb の電子数 を減少させる必要があると考える。 謝辞 本研究の一部は,東京大学情報基盤センターのスーパーコンピュータ Reedbush-U を利用して 実施された。
117 付録 A AIMD の計算の詳細 固液界面で,ある種の化学反応が起きることが既知であるとき,その化学反応に関係する物 性値,例えば自由エネルギーを定量的に評価できる計算条件を設定しなければならない。特に 周期境界条件のもとで,固体部分をスラブとして取り扱い,固体と固体の間に液体部分を挿入 する場合,固体スラブの厚み (結晶であるなら,積層数) ,液体部分の厚み,さらに液体の濃度 に関連する固体スラブの表面に垂直な方向の格子ベクトルの長さを決める必要がある。原理的 には,これらの計算条件を変化させることによって得られる物性値の収束性を注意深く調べ, 定量性のある物性値の予測値が決められる。 本研究の場合には,固液界面で化学反応が起きることが既知ではなく,化学反応が起きない ことも起こり得る。事実,本研究で示されたように 50 ps の AIMD 時間の中では,Pb と H2SO4(aq)の界面で化学反応は観察されないことがわかった。本研究では,このような化学反応 が未知の系の計算条件 (固体スラブの厚みと液体スラブの厚み) を決めるために,上述の方法か らは外れるが,真空層スラブ模型の表面エネルギーを使用した。固体-液体スラブと固体-真 空スラブは,全く異なる系であり,このような代替的な固体-真空スラブの利用が正しいとい う保証はない。しかしながら,化学反応が未知の系で計算機実験を実施するための計算条件の 決定には,最も効率的な手法であると考える。 最初に 6 層積層した Pb(111),Pb(100),Pb(110)スラブを使って,真空層の厚さを変化させるこ とによって表面エネルギーを計算した。その結果,1 nm の真空層の厚さで十分収束した表面エ ネルギーが得られた。SO42–イオンは直径が約 0.5 nm の大きさを持つため,1 nm の液体スラブの 厚みでは不十分と考えられた。それゆえ,液体スラブの厚みは,1 nm の 2 倍の 2 nm と決められ た。 次にスラブ間隔を 2 nm に固定して,スラブ積層数を変化させることによって表面エネルギー を計算した。Fig. A1 に示されるように表面エネルギーは 10 層積層したときでさえ,十分収束し なかった。5d 電子を含んだ電子状態計算から得られた表面エネルギーは,5d 電子を含まない電 子状態計算から得られたものよりわずかに低エネルギー側にシフトした。7 層以上の積層数で は,上下の表面 3 層のみを緩和させた表面エネルギーが計算された。例えば,8 層の積層数のス ラブでは,中央の 2 層の原子の位置は固定され,上表面 3 層と下表面 3 層の原子がそれぞれ緩 和される。スラブの中央の原子を固定した表面エネルギーは,すべての原子を緩和させたもの と比べて,積層数の増加にとともにやや揺らぎが大きくなった。本研究で得られた表面エネル ギーの結果は,Li らの 5d 電子を含まない PAW–PBEsol の計算条件での Pb スラブの積層数に対 する表面エネルギーの折れ線図と比べて,表面エネルギーの大きさは異なるが,傾向はよく似 ていた[26]。Li らの結果から,よく収束した Pb の表面エネルギーを得るためには少なくとも 26 層必要であることがわかる[26]。本研究では,Pb と H2SO4(aq)の界面での化学反応が起きるかど うかを調べることを目的としており,真空層スラブ模型でのスラブ積層数に対する表面エネル ギーの収束性を調べることを目的としていない。そのため,私は Pb と H2SO4(aq)の界面での AIMD 計算の計算条件として,真空層スラブ模型での表面エネルギーが十分収束していないに もかかわらず,Pb 原子 6 層の積層数を選んだ。 スーパーセルの a 軸と b 軸の長さは,隣のスーパーセル内の SO42–イオンからの離隔を考慮し て,特定の Pb 表面を z 軸方向に選んだ単位胞の整数倍を選び,約 1 nm となるように設定した ((100)面については 2×2,(110)面については 2×3,(111)面については 3×3 倍) 。3 種類の単位 胞はすべて直方晶であるので,単純に a 軸方向の長さと b 軸方向の長さを掛け算することによ って,スーパーセルの表面積が計算された (Table 1 参照) 。 さらに Pb と H2SO4(aq)の界面での AIMD 計算中の電子状態計算では,6s6p 軌道の電子のみが 価電子として考慮した。Pb は sp 金属であるため,遷移金属と比べて結合エネルギーが小さく [27],Fig. 1 の第一近接の半値幅が示すように,300 K の温度で Pb スラブはかなり揺らぐ。それ ゆえ,遷移金属表面の真空層スラブ模型の 0 K DFT 計算でよく使われるようなスラブの中心部 を固定する制約条件を使用せず,バルク水溶液の AIMD 計算で使用される計算条件であるスー
118
パーセル内の全原子の重心の運動エネルギーを 0 に固定する計算条件が使われた。AIMD 計算 と同じ計算条件で,表面エネルギーが計算された。
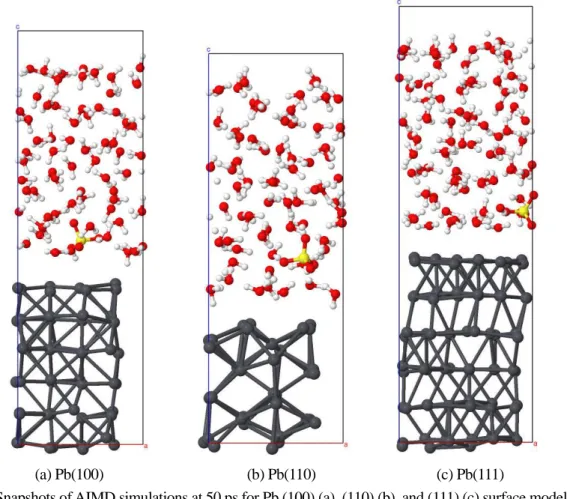
Figure A2 は,H2SO4(aq)をともなった 3 種類の Pb 低指数表面の AIMD 計算の MD 時間 50 ps
後のスナップショットを示す。
(a) PAW–PBE–D3(5d106s26p2) (b) PAW–PBE–D3(6s26p2)
Fig. A1 Surface energy convergence in function of slab thickness for (100), (110), and (111) surfaces with
vacuum thickness of 2 nm obtained using 5d106s26p2 electrons (left panel) and 6s26p2 electrons (right panel).
Solid lines indicate results relaxed for all atoms, and dotted ones are results relaxed for three layers of both top and bottom sides of surface.
(a) Pb(100) (b) Pb(110) (c) Pb(111)
Fig. A2 Snapshots of AIMD simulations at 50 ps for Pb (100) (a), (110) (b), and (111) (c) surface models with
H2SO4(aq).
Gray, yellow, red, and white spheres indicate Pb, S, O, and H atoms, respectively. Rectangles show supercells used in this study.
2 3 4 5 6 7 8 9 10 350 400 500 600 (110) (100) (111) S u rf ace en er g y ( m J m -2) Slab thickness of Pb 450 550 2 3 4 5 6 7 8 9 10 350 400 500 600 (110) (100) (111) S u rf ace en er g y ( m J m -2) Slab thickness of Pb 450 550
119 参考文献
[1] D. Pavlov, Lead-Acid Batteries Science and Technology 2nd edit. Elsevier US 2017. [2] H. A. Catherino, F. F. Feres, and F. Trinidad, J. Power Sources 129, 113–120 (2004). [3] A. Kirchev, F. Mattera, E. Lemaire, and K. Dong, J. Power Sources 191, 82–90 (2009). [4] H. Karami and R. Asadi, J. Power Sources 191, 165–175 (2009).
[5] G. Kresse and J. Hafner, Phys. Rev. B 47, 558−561 (1993). [6] G. Kresse and J. Hafner, Phys. Rev. B 49, 14251−14269 (1994). [7] G. Kresse and J. Furthmüller, Phys. Rev B 54, 11169−11186 (1996). [8] G. Kresse and J. Furthmüller, Comput. Mater. Sci. 6, 15−50 (1996). [9] P. E. Blöchl, Phys. Rev. B 50, 17953−17979 (1994).
[10] G. Kresse and D. Joubert, Phys. Rev. B 59, 1758−1775 (1999).
[11] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865−3868 (1996).; 78, 1396(E) (1997). [12] S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, J. Chem. Phys. 132, 154104 (2010).
[13] T. Bučko, J. Hafner, S. Lebègue, and J. G. Ángyán, J. Phys. Chem. A 114, 11814–11824 (2010). [14] S. Nosé, J. Chem. Phys. 81, 511−519 (1984).
[15] W. G. Hoover, Phys. Rev. A 31, 1695−1697 (1985). [16] F. Birch, Phys. Rev. 71, 809–824 (1947).
[17] D. W. Green and R. H. Perry, Perry’s Chemical Engineers’ Handbook, 7th edit. McGraw-Hill Professional 1997. TABLE 2-201.
[18] D.Yu and M. Scheffler, Phys. Rev. B 70, 155417 (2004).
[19] W. Li, L. Huang, R. G. S. Pala, G.-H. Lu, F. Liu, J. W. Evans, and Y. Han, Phys. Rev. B 96, 205409 (2017).
[20] H. P. Klug, J. Am. Chem. Soc. 68, 1493–1494 (1946).
[21] Y. K. Vohra and A. L. Ruoff, Phys. Rev. B 42, 8651–8654 (1990).
[22] C. Kittel, Introduction to Solid State Physics, 8th edit. Hoboken, NJ John Wiley & Sons, Inc., 2005. p. 8.
[23] I. Galanakis, N. Papanikolaou, and P.H. Dederichs, Surf. Sci. 511, 1–12 (2002). [24] C. Bombis, A. Emundts, M. Nowicki, and H. P. Bonzel, Surf. Sci. 511, 83-96 (2002). [25] Y. Kubota, Unpublished work.
[26] W. Li, L. Huang, R. G. S. Pala, G.-H. Lu, F. Liu, J. W. Evans, and Y. Han, Phys. Rev. B 96, 205409 (2017).