ミルセラ注シリンジ 25 μg ミルセラ注シリンジ 50 μg ミルセラ注シリンジ 75 μg ミルセラ注シリンジ 100 μg ミルセラ注シリンジ 150 μg ミルセラ注シリンジ 200 μg ミルセラ注シリンジ 250 μg
[腎性貧血]
第 2 部 CTD の概要(サマリー)
2.5 臨床に関する概括評価(臨床概括評価)
中外製薬株式会社
略語一覧
略語 英名 和名
ADL Activities of Daily Living 日常生活動作 Alb Albumin アルブミン ALT
(GPT)
Alanine aminotransferase
(Glutamic pyruvic transaminase)
アラニンアミノトランスフェラーゼ
(グルタミン酸ピルビン酸トランスアミナーゼ)
ALP Alkaline phosphatase アルカリフォスファターゼ
APTT Activated partial thromboplastin time 活性化部分トロンボプラスチン時間 AST
(GOT)
Aspartate aminotransferase
(Glutamic oxaloacetic transaminase)
アスパラギン酸アミノトランスフェラーゼ
(グルタミン酸オキサロ酢酸トランスアミナーゼ)
AUC Area under the concentration-time curve 血清中濃度-時間曲線下面積 AUCinf
(AUC0-∞)
Area under the concentration-time curve from time zero to infinity
(投与時から無限大時まで外挿した)血清中濃度-
時間曲線下面積 AUClast Area under the concentration-time curve from
time zero to the last measurable concentration
(投与時から最終測定時点までの)血清中濃度-時 間曲線下面積
AUE Area under the effect-time curve 効果-時間曲線下面積
BA Bioavailability 生物学的利用率(バイオアベイラビリティ)
Baso Basophil granulocyte 好塩基球 BC-AUE Baseline-corrected under the effect-time
curve ベースライン補正した効果-時間曲線下面積 BL Baseline Hb levels ベースラインヘモグロビン濃度
BMI Body mass index ボディマス指数
BNP Brain natriuretic peptide 脳性ナトリウム利尿ポリペプチド BUN Blood urea nitrogen 血中尿素窒素
Ca Calcium カルシウム
CAPD Continuous ambulatory peritoneal dialysis 連続携行式腹膜透析 Ccr Creatinine clearance クレアチニンクリアランス CHMP Committee for Medicinal Products for Human
Use
医薬品委員会(欧州医薬品審査庁において医薬品の 科学的評価を担当する医薬品委員会)
CI Confidence interval 信頼区間 CKD Chronic kidney disease 慢性腎臓病 Cl Chlorine クロール
CL Clearance(Dose/AUC0-∞) クリアランス(用量/AUC0-∞)
CL/F Clearance/Bioavailability(Dose/AUC0-∞) 皮下投与時のクリアランス(用量/AUC0-∞) Cav Average serum drug concentration 平均血清中薬物濃度
Cmax Maximum serum drug concentration 最高血清中薬物濃度 Cmin Minimum serum drug concentration 最低血清中薬物濃度 Cr,CRE Creatinine クレアチニン CRDAC Cardiovascular and Renal Drugs Advisory
Committee 心血管用薬および腎臓用薬諮問委員会 CRF Case report form 症例報告書
CRP C-reactive protein C反応性蛋白 CTR Cardio-thoracic ratio 心胸郭比 CVD Cardiovascular disease 心血管系疾患 EBM Evidence Based Medicine 根拠に基づいた医療
EBPG European Best Practice Guideline 欧州版最良診療ガイドライン EDTA European Dialysis and Transplantation
Association 欧州透析移植学会 Emax Maximum effect 最大効果(最大増加量)
EMEA European Medicines Evaluation Agency 欧州医薬品審査庁
EOIT End of initial treatment 投与開始日から投与量変更前までの期間 Eosino Eosinophil granulocyte 好酸球
EPO Erythropoietin エリスロポエチン EPO-β Epoietin beta エポエチン ベータ
略語 英名 和名
ERBP European Renal Best Practice 欧州版腎臓診療指針 ESA Erythropoiesis stimulating agent 赤血球造血刺激因子製剤 ESKD End-stage kidney disease 末期腎不全
FAS Full analysis set 最大の解析対象集団 FDA Food and Drug Administration 米国食品医薬品局
Fe Iron 鉄
GCP Good Clinical Practice 医薬品の臨床試験の実施に関する基準 GFR Glomerular filtration rate 糸球体濾過量
γ-GTP γ-Glutamyltranspeptidase γ-グルタミルトランスペプチダーゼ
hANP Human atrial natriuretic peptide ヒト心房性ナトリウム利尿ポリペプチド Hb Hemoglobin ヘモグロビン
HCG Human chorionic gonadotropin ヒト絨毛性ゴナドトロピン HD Hemodialysis 血液透析
Ht Hematocrit ヘマトクリット
intact-PTH Intact parathyroid hormone PTHインタクト(完全分子型)
ITT Intention–to–treat – i.v. Intravenous 静脈内 IVST Intraventricular septum thickness 心室中核壁厚 JSDT Japanese Society of Dialysis Treatment 日本透析医学会 JSN Japanese Society of Nephrology 日本腎臓学会 K Potassium カリウム
KDOQI Kidney Disease Outcomes Quality Initiative 腎臓病患者の予後改善機構 Kt/V - 透析量を表す指標
LDH Lactate dehydrogenase 乳酸脱水素酵素 LsMeans Least-squares means 最小二乗平均 LVDd Left ventricular end-diastolic diameter 左室拡張期径 LVDs Left ventricural end-systolic diameter 左室収縮期径 LVMI Left ventricular mass index 左室心筋重量係数 LVPWT Left ventricular posterior wall thickness 左室後壁厚 Lymph Lymphocyte リンパ球
M&S Model&Simulation モデル&シミュレーション Max Maximum 最大値
MCH Mean corpuscular hemoglobin 平均赤血球血色素量 MCHC Mean corpuscular hemoglobin
concentration 平均赤血球血色素濃度 MCV Mean corpuscular volume 平均赤血球容積 MedDRA Medical Dictionary for Regulatory Activities ICH国際医薬用語集 Min Minimum 最小値
Mono Monocyte 単球
mPEG Methoxy polyethylene glycol メトキシポリエチレングリコール MRT Mean residence time 平均滞留時間
Na Sodium ナトリウム Neut Neutrophil granulocyte 好中球 NKF National Kidney Foundation 米国腎臓財団
NYHA New York Heart Association ニューヨーク心臓協会 P Phosphorus 無機リン
PD Pharmacodynamic 薬力学的反応 PK Pharmacokinetics 薬物動態 PK/PD Pharmacokinetic/Pharmacodynamic 薬物動態/薬力学 PLT Platelet 血小板
PPS Per protocol set 治験実施計画に適合した対象集団 PSUR Periodic Safety Update Report 定期的安全性最新報告
PT Preferred term 基本語(MedDRA)
略語 英名 和名
PT Prothrombin time プロトロンビン時間 PTA Percutaneous transluminal angioplasty 経皮的血管形成術 QOL Quality of Life 生活の質
RBC Red blood cell 赤血球
RC-Emax Relative change of Emax 最大効果のベースラインに対する比率 Ret Reticulocyte 網状赤血球
rHuEPO Recombinant human erythropoietin 遺伝子組換えヒトエリスロポエチン RIP Radioimmunoprecipitation 放射免疫沈降法
s.c. Subcutaneous 皮下 SD(Std) Standard deviation 標準偏差 SE Standard error 標準誤差 SF-36 Short-Form 36-Item Health Survey SF健康調査票
SLE Systemic lupus erythematosus 全身性エリテマトーデス t1/2 Elimination half-life 消失半減期
T-Bil Total bilirubin 総ビリルビン T-cho Total cholesterol 総コレステロール TIBC Total iron binding capacity 総鉄結合能
tmax Time to maximum serum concentration 最高血清中濃度到達時間 TP Total protein 総蛋白
TSAT Transferrin saturation トランスフェリン飽和度 UA Uric acid 尿酸
Vd,β/F Volume of distribution based on the terminal
phase 消失相に基づく皮下投与時の分布容積 Vss,Vd,ss Volume of distribution at steady state 定常状態での分布容積
WBC White blood cell 白血球
目次
頁
2.5 臨床に関する概括評価 ... 8
2.5.1 製品開発の根拠 ... 8
2.5.1.1 背景 ... 8
2.5.1.1.1 慢性腎臓病の疫学 ... 8
2.5.1.1.2 慢性腎臓病に伴う腎性貧血 ... 9
2.5.1.1.3 腎性貧血に対するESAの治療 ... 9
2.5.1.1.4 腎性貧血治療のガイドライン ... 11
2.5.1.2 国内及び海外における開発の経緯 ... 14
2.5.1.2.1 国内における開発の経緯 ... 14
2.5.1.2.1.1 臨床データパッケージ ... 17
2.5.1.2.1.2 治験相談 ... 21
2.5.1.2.2 試験デザインに関する考察 ... 23
2.5.1.2.3 海外における開発及び承認状況 ... 25
2.5.1.3 医薬品の臨床試験実施に関する基準(GCP)遵守 ... 28
2.5.1.3.1 重大なGCP違反 ... 28
2.5.1.3.2 当該症例の取扱い ... 28
2.5.1.3.3 臨床データパッケージに使用した症例の内訳 ... 32
2.5.2 生物薬剤学に関する概括評価 ... 33
2.5.2.1 製剤 ... 33
2.5.2.2 固定用量の適切性 ... 34
2.5.3 臨床薬理に関する概括評価 ... 35
2.5.3.1 健康成人における薬物動態 ... 36
2.5.3.2 慢性腎臓病患者における薬物動態 ... 36
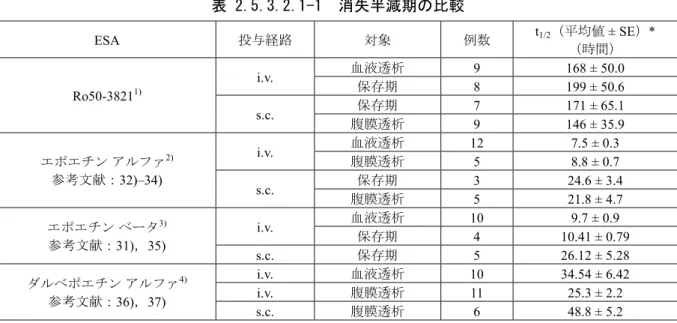
2.5.3.2.1 消失半減期 ... 37
2.5.3.2.2 暴露量の用量比例性 ... 38
2.5.3.2.3 長期投与における血清中Ro50-3821濃度... 38
2.5.3.3 病期間での薬物動態及び薬力学的反応の比較 ... 39
2.5.3.3.1 病期間の薬物動態の比較 ... 39
2.5.3.3.2 病期間の薬力学的反応の比較 ... 40
2.5.3.3.3 病期間での薬物動態及び薬力学的反応のまとめ ... 40
2.5.3.4 投与経路間の薬物動態及び薬力学的反応の比較 ... 40
2.5.3.4.1 投与経路間の薬物動態の比較 ... 40
2.5.3.4.2 投与経路間の薬力学的反応の比較 ... 41
2.5.3.4.3 投与経路間の薬物動態及び薬力学的反応のまとめ ... 41
2.5.3.5 排泄,代謝 ... 42
2.5.3.5.1 排泄 ... 42
2.5.3.5.2 代謝 ... 42
2.5.3.6 薬物動態に影響する因子 ... 42
2.5.3.7 母集団薬物動態/薬力学解析に基づくModel&Simulation ... 43
2.5.4 有効性の概括評価 ... 46
2.5.4.1 対象患者及び患者背景 ... 48
2.5.4.2 初期投与期における貧血改善効果 ... 48
2.5.4.2.1 血液透析における貧血改善効果 ... 49
2.5.4.2.2 保存期における貧血改善効果 ... 50
2.5.4.3 維持投与期における貧血改善維持効果 ... 54
2.5.4.3.1 血液透析における貧血改善維持効果 ... 55
2.5.4.3.2 保存期における貧血改善維持効果 ... 56
2.5.4.3.3 腹膜透析における貧血改善維持効果 ... 60
2.5.4.4 エポエチン ベータとの比較 ... 61
2.5.4.4.1 血液透析における切替維持効果の比較(JH20876) ... 61
2.5.4.4.2 保存期における貧血改善維持効果の比較(JH20565) ... 63
2.5.4.4.3 保存期患者における貧血改善維持効果の比較(JH22757) ... 66
2.5.4.4.4 海外におけるESAとの貧血改善維持効果の比較 ... 68
2.5.4.5 有効性に影響する因子 ... 69
2.5.4.6 効能・効果,用法・用量 ... 69
2.5.4.6.1 効能・効果 ... 69
2.5.4.6.2 用法・用量 ... 69
2.5.4.6.2.1 初期投与期における用法・用量 ... 69
2.5.4.6.2.2 維持投与期における用法・用量 ... 71
2.5.4.6.2.3 rHuEPO製剤からの切替初回用量 ... 73
2.5.4.6.2.4 2週に1回から4週に1回への切替用量 ... 76
2.5.5 安全性の概括評価 ... 77
2.5.5.1 患者背景及び曝露量 ... 80
2.5.5.2 有害事象 ... 80
2.5.5.2.1 比較的よくみられる有害事象 ... 81
2.5.5.2.2 部分集団における有害事象 ... 82
2.5.5.2.3 時期別の有害事象 ... 82
2.5.5.2.4 死亡に至った有害事象 ... 83
2.5.5.2.5 その他の重篤な有害事象 ... 84
2.5.5.2.6 治験中止に至った有害事象 ... 85
2.5.5.2.7 エポエチン ベータとの比較 ... 85
2.5.5.2.8 有害事象とHb濃度との関係 ... 88
2.5.5.2.9 重要な有害事象の発現 ... 89
2.5.5.2.10 臨床検査値,バイタルサイン等 ... 90
2.5.5.3 海外臨床試験における安全性 ... 92
2.5.5.4 海外における市販後安全性報告 ... 93
2.5.5.5 有害事象の予防,軽減,管理方法 ... 94
2.5.5.6 過量投与に対する対応,依存性,反跳現象,乱用を誘発する可能性,又はそれ らのデータの欠如 ... 95
2.5.6 ベネフィットとリスクに関する結論 ... 96
2.5.6.1 ベネフィット ... 98
2.5.6.2 リスク ... 104
2.5.7 参考文献 ... 106
2.5 臨床に関する概括評価 2.5.1 製品開発の根拠 2.5.1.1 背景
Ro50-3821(エポエチン ベータ ペゴル(遺伝子組換え))(以下,本剤とする)は,エポエ
チン ベータ(遺伝子組換え)に1分子の直鎖メトキシポリエチレングリコール(mPEG)分子 を化学的に結合させ合成した。本剤は,遺伝子組換えヒトエリスロポエチン(rHuEPO)製剤 やダルベポエチン アルファ等の既存の赤血球造血刺激因子製剤(Erythropoiesis Stimulating Agent:ESA)に比べて静脈内及び皮下投与いずれの投与経路においても大幅に消失半減期
(t1/2)が延長され1),海外では月1回の用法で承認されている新しい ESA である(1.6.3,1.6.4 参照)。
通常,保存期及び腹膜透析患者の通院頻度は2~4週に1回であるが,rHuEPO製剤による適切 な貧血治療を実施するためには1~2週に1回の投与が必要であるため,貧血治療が不十分であ る場合が多い2),3)。本剤は,患者の通院頻度に合わせ静脈内及び皮下投与いずれの投与経路に おいても4週に1回の投与で適切な貧血治療が可能となるため通院負担が軽減され,また,治療 コンプライアンスが向上することにより,QOL や心機能の改善,透析導入の遅延等,特に保 存期患者における治療効果の向上に大きく寄与するものと考えられる。
一方,血液透析患者の rHuEPO 製剤による貧血治療は,通常,透析施行時に週に2~3回の投 与で,生涯に亘って治療を継続する必要がある。本剤による貧血維持治療では,投与頻度が4 週に1回となり,rHuEPO 製剤に対して1/12~1/8の投与頻度となることから,医療過誤や感染 リスクの低減,医療業務及び医療廃棄物などの医療コストの削減が見込まれ,血液透析医療に 貢献できるものと考えられる。
これらのことから,本剤による新しい貧血治療は,医療現場の多様なニーズを満たすととも に,慢性腎臓病(CKD)の医療の発展に大きく貢献するものと考えられ,本剤の開発に至っ た。
2.5.1.1.1 慢性腎臓病の疫学
近年,CKD は日本のみならず世界中で増加し続ける末期腎不全(End-stage kidney disease:
ESKD)の予備群として注目されている。CKDは,蛋白尿などの腎臓の障害若しくは糸球体濾
過量(GFR)が60 mL/min/1.73 m2未満の腎機能低下が3カ月以上続く状態と定義され,その病 期(ステージ)は表 2.5.1.1.1-1に示す基準により分類されている4)。
2008年5月に開催された第51回日本腎臓学会学術総会において,国内の CKD(尿蛋白陽性又
はGFR 60 mL/min/1.73 m2未満)患者数は約1,330万人(全人口の12.9%)であり,ESKDや心・
血管系疾患(Cardiovascular disease:CVD)のリスクが高まるとされるGFR 50 mL/min/1.73 m2 未満の患者数は約591万人(全人口の5.7%)であることが報告された。また,日本透析医学会
(Japanese Society of Dialysis Treatment:JSDT)が行っている2008年末時点の統計調査5)では,
透析患者数は約28.3万人と報告され,年間の新規導入患者数は約3.8万人,死亡患者数は約2.7 万人と年間に約1万人が増加しており,今後も同程度の増加が続くものと予想されている。透 析患者の死亡原因は,心不全(24.0%),脳血管障害(8.6%),心筋梗塞(4.2%)などを含め た CVD が最も多く,次いで感染症(20.0%)であった5)。また,透析患者の原疾患は,慢性糸 球体腎炎(39.0%),糖尿病性腎症(34.2%),腎硬化症(6.8%),多発性嚢胞腎(3.4%)の 順で多く,特に糖尿病性腎症の増加が著しい。この傾向は今後も続くと考えられており,世界 的な傾向と一致している。
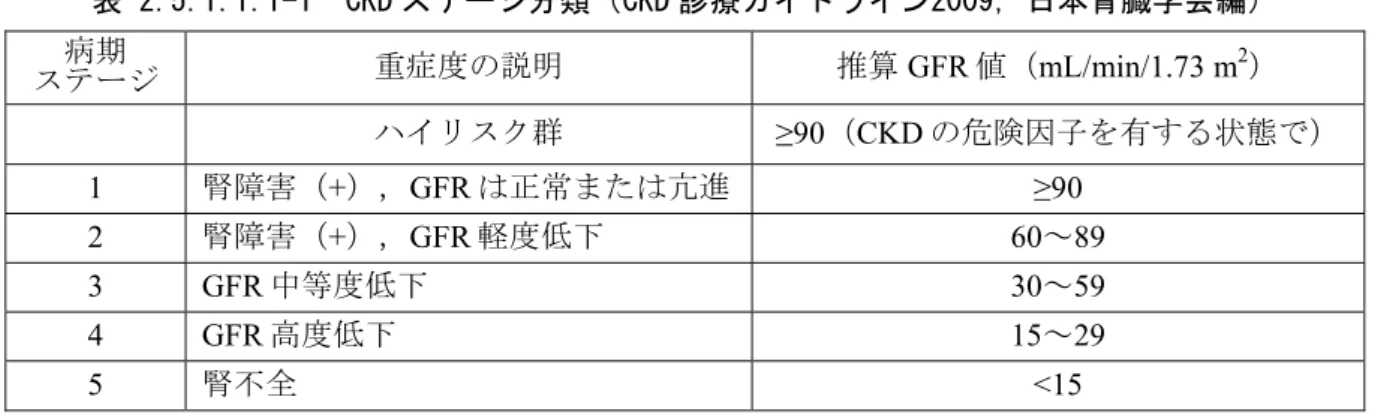
表 2.5.1.1.1-1 CKD ステージ分類(CKD 診療ガイドライン2009,日本腎臓学会編)
ステージ 病期 重症度の説明 推算GFR値(mL/min/1.73 m2) ハイリスク群 ≥90(CKDの危険因子を有する状態で)
1 腎障害(+),GFRは正常または亢進 ≥90 2 腎障害(+),GFR軽度低下 60~89
3 GFR中等度低下 30~59
4 GFR高度低下 15~29
5 腎不全 <15
2.5.1.1.2 慢性腎臓病に伴う腎性貧血
CKD は腎障害を示す所見や腎機能低下が慢性的に続く状態のことである。腎性貧血は CKD のステージ3以前からも出現し,ステージが進行するに従い発現頻度が急激に増加して,
ESKDとなるステージ5の透析期では最終的に大半の患者が腎性貧血を併発する6)。
JSDT による2008年版「慢性腎臓病患者における腎性貧血治療のガイドライン」7)において,
腎性貧血の主因は腎障害に伴うエリスロポエチン(EPO)の産生低下であり,これ以外に貧血 の原因疾患が認められないときに初めて診断されると述べられている。その診断の詳細は表 2.5.1.1.2-1のとおりである。
透析患者の死亡原因の多くは心不全,脳血管障害,心筋梗塞等の CVD であり,また,CKD に合併する腎性貧血は CVD の独立した危険因子と考えられていることから,腎性貧血の治療 を行うことによりCVDの発症・進展を予防し,CKDの増悪を防ぐことが重要であると考えら れている8)。
表 2.5.1.1.2-1 腎性貧血の診断と基準
腎性貧血の頻度が急激に増加する糸球体濾過量(GFR)低下の目安は,血清クレアチニン
(Cr)値 ≥ 2 mg/dLまたはクレアチニンクリアランス(Ccr)値 < 20~35 mL/min程度(CKD stage 4~5)である。糖尿病性腎症では,非糖尿病性腎症患者より早期に腎性貧血が出現する といわれており,その目安はCcr値 < 45 mL/min程度である。
しかし,これ以上の GFR を有する場合でも腎性貧血の存在は否定できない。「腎性貧血は内 分泌疾患」であるとの観点から,診断には,腎障害に伴う腎での EPO 産生の低下以外の貧血 の原因疾患が否定されていなければならない。しかし,GFR の低下が上記の範疇にあり,他 に貧血の原因(特に鉄欠乏性貧血など)が判明しない場合にはほぼ腎性貧血と診断してよ い。
2.5.1.1.3 腎性貧血に対する ESA の治療
(1) 赤血球造血刺激因子製剤(ESA)
CKD における腎性貧血は,造血ホルモンである EPO の相対的欠乏により発症すると考えら れており,EPO 受容体を刺激し赤血球造血作用を有する ESA がその治療の第一選択薬として 位置づけられている。国内では,rHuEPO 製剤としてエポエチン ベータ,エポエチン アルフ ァ及びエポエチン カッパ[エポエチン アルファ後続1],更に持続型 ESA としてダルベポエ チン アルファが販売されている。
エポエチン ベータは,遺伝子組換え技術によってヒト EPO 遺伝子を組み込んだチャイニー ズハムスター卵巣細胞株により産生されるアミノ酸165個を有する分子量約30,000の糖たん白 質であり,天然型ヒト EPOと基本的構造に差異がないことが確認されている。エポエチン ベ
ータ製剤は中外製薬が開発し,エポジン注9)として「腎性貧血」,「自己血貯血」及び「未 熟児貧血」の効能が承認され臨床使用されている。エポエチン ベータの腎性貧血での投与経 路ごとの適応を表 2.5.1.1.3-1に示した。エポエチン ベータは血液透析患者では静脈内投与のみ,
腹膜透析及び保存期患者では静脈内投与,皮下投与ともに承認されている。海外では,ロシュ 社がエポエチン ベータをNeoRecormonとして「腎性貧血」,「がん化学療法による貧血」,
「自己血貯血」及び「未熟児貧血」の適応症で販売している。
また,エポエチン アルファは,エポエチン ベータ同様のアミノ酸配列を持ち,有効性,安 全性も類似している10),52)。ダルベポエチン アルファは,エポエチン アルファの5カ所のアミノ 酸を改変することにより糖鎖を増やして t1/2を延長した持続型ESAであり,「腎性貧血」の効 能・効果として承認されている53)。
表 2.5.1.1.3-1 腎性貧血におけるエポエチン ベータの効能・効果及び投与経路
効能・効果 腎性貧血
対象 血液透析 腹膜透析 透析導入前
(保存期)
投与経路 静脈内投与 ○ ○ ○
皮下投与 - ○ ○
(2) ESAによる治療効果
透析患者の腎性貧血に対して,従来,輸血以外に主たる治療方法はなかったが,rHuEPO 製 剤は著しい貧血の改善効果を示し,労作意欲の低下,活動性の低下,易疲労,動悸,息切れ,
立眩み,頭痛,食欲の低下,性欲減退等の貧血による自覚症状が確実に改善・消失するととも に,輸血量の減少と輸血に伴う副作用,鉄沈着症の激減が認められ,身体及び精神両面からの QOL の向上をもたらした。また,CVD による死亡患者の減少,予後の改善等,多くの治療効 果が報告されている12)。
保存期患者に対しては,ESA 治療により透析導入の遅延や腎不全の進行抑制(腎保護作用)
が国内外で報告されている13),14)。保存期患者での ESA による腎機能以外への効果は,基本的 に透析患者と同様と考えられ,心機能をはじめとした様々な臓器機能の改善効果,日常生活動 作(ADL)やQOLの向上が確認されている15),16)。
近年,心不全,腎不全,貧血が相互に影響し合う諸事実が明らかになり,心・腎・貧血症候 群の概念が提唱されている17)。腎臓に対する貧血の影響として,貧血が高度になるほど CKD 患者では腎死率が高くなるが6),ESA により貧血を積極的に治療することにより,腎機能の進 行悪化が抑制されると報告されている13),14)。また,慢性的な貧血の存在は心臓の仕事量を増や し,その結果として心肥大を招き心不全の発症をもたらすが6),ESA により貧血を改善するこ とにより,心拍数,心拍出量の有意な減少,左室心筋重量係数(Left Ventricular Mass Index:
LVMI)の改善,最大酸素消費量の増大等,心機能が改善することが報告されている18)–21)。
以上のことから,CKD 患者においては,早期より腎性貧血を積極的に改善することが透析 導入時期を遅らせ,更には保存期及び透析期を含めた患者の CVD による死亡を減少させるこ とが明らかとなり,腎性貧血の発症早期より積極的に治療を行うことが重要であるとされてい る。
2.5.1.1.4 腎性貧血治療のガイドライン
(1) 国内外における腎性貧血治療のガイドライン
国内では2004年に JSDT より「慢性血液透析患者における腎性貧血治療のガイドライン」22) が示されたが,腹膜透析及び保存期患者については,国内のガイドラインが存在しなかった。
その後,日本腎臓学会(Japanese Society of Nephrology:JSN),日本腹膜透析研究会,日本小 児腎臓病学会の共同で検討が重ねられ,2008年10月に JSDTより血液透析患者に加え腹膜透析 患者,保存期患者及び小児患者に対する「慢性腎臓病患者における腎性貧血治療のガイドライ ン」7)が公表され,腹膜透析及び保存期患者における ESA 療法の目標ヘモグロビン(Hb)濃 度が新たに示された。更に,日本腎臓学会からは2009年に CKD 診療ガイドライン8)が公表さ れ,JSDTと同様な腎性貧血の治療方針が示された。
一方,海外において,腎性貧血の治療ガイドラインはEvidence Based Medicine(EBM)を背 景に,米国では1997年に National Kidney Foundation(NKF)より Kidney Disease Outcomes Quality Initiative(KDOQI) ガ イ ド ラ イ ン が , 欧 州 で は1999年 に European Dialysis and Transplantation Association(EDTA)より European Best Practice Guideline(EBPG)が示された。
その後それぞれのガイドラインで改訂が重ねられ,KDOQI は2007年版23),EDTA は2009年に ポジションステートメントとしてEuropean Renal Best Practice(ERBP)24)と2010年にERBPに 対するポジションステートメント54)を公表した。
(2) ESA療法の投与開始基準と目標Hb濃度
国内外のガイドラインにおける目標 Hb 濃度(表 2.5.1.1.4-1)は,海外のガイドラインでは CKD 患者を一括りとしているのに対して,国内のガイドラインでは血液透析患者とそれ以外
(保存期及び腹膜透析患者)に分けて示されている。血液透析患者での目標値は10~11 g/dL であり,12 g/dLを超えた場合は減量・休薬,また,若年者では11~12 g/dLであり,13 g/dLを 超えた場合は減量・休薬とされている。保存期及び腹膜透析患者での目標値は11 g/dL 以上で あり,13 g/dLを超えた場合は減量・休薬,重篤な CVD 患者は12 g/dLを超えた場合,減量・
休薬とされている。ESA の投与開始基準は,血液透析患者では10 g/dL 未満,保存期及び腹膜 透析患者では11 g/dL未満に設定されている。血液透析患者は保存期及び腹膜透析患者での Hb 濃度に比べ低く設定されているが,その理由は血液透析の除水により生じる血液濃縮の結果,
透析後のHb濃度が1~3 g/dL上昇するためである。
表 2.5.1.1.4-1 国内外のガイドラインの目標 Hb 濃度 JSDT(2008年)
JSN*(2009年) KDOQI(2007年) ERBP(2009年,2010年)
<血液透析患者>
目標:10~11 g/dL
減量・休薬:12 g/dLを超えた場合
(若年者:11~12 g/dL,13 g/dLを超え た場合,減量・休薬)
<保存期患者・腹膜透析患者>
目標:11 g/dL以上
減量・休薬:13 g/dLを超えた場合
(重篤な CVD患者:12 g/dLを超えた場 合,減量・休薬)
<CKD患者>
目標:11~12 g/dL
13 g/dLを超えるべきではない
<CKD患者>
目標:11~12 g/dL
13 g/dLを超えるべきではない
(脳卒中の既往を有するⅡ型糖 尿病患者:10~12 g/dL**)
*:JSN2009年版は保存期患者のみ
**:ポジションステートメント2010年版のリコメンデーション
JSDTのガイドラインでは,保存期及び腹膜透析患者での国内で実施された ESAの試験成績 より,Hb 濃度11 g/dL 以上での安全性には問題のないこと,Hb 濃度を11 g/dL 未満に比べて
11 g/dL以上に維持することにより QOLや心機能改善の効果が認められたことから25),26),海外
のガイドラインと同様に,国内でも目標Hb濃度を11 g/dL以上に設定することは妥当とされて いる。また,Hb濃度の上昇によりQOLの向上やLVMIの改善などの有用性が期待されること から,これらの試験で除外された重篤な CVD を有する患者を除いては,目標 Hb 濃度を
12 g/dL以上に設定することも可能と考えられている。
一方,保存期患者を対象に海外で実施された CHOIR試験27)において,rHuEPO製剤を投与し て目標 Hb濃度を13.5 g/dLとした群(高Hb濃度群)と11.3 g/dLとした群を比較した場合,高 Hb 濃度群において重篤かつ死亡のおそれのある心・血管系疾患が増加したとの結果が報告さ れ,米国FDAは2006年11月16日付けで推奨の目標Hb濃度の範囲「Hb濃度10~12 g/dLを維持 すること」を遵守するよう注意喚起を行い,2007年3月9日に本内容を添付文書に記載するよう 指示した。
その後,CHOIR試験の結果を含むメタアナリシス結果が発表され,高いHb濃度群(12 g/dL 以上)では,死亡やシャント閉塞,血圧管理不良のリスクが有意に高まると報告された28)。こ れらの新しいエビデンスの評価結果から KDOQI ガイドラインの Hb 濃度の上限値に関する記 載は,2006年版の「Hb 濃度を13 g/dL 以上にする根拠はない」から2007年版では「Hb 濃度
13 g/dL を超えるべきではない」と変更された。また,2007年9月には FDA の諮問委員会であ
るCardiovascular and Renal Drugs Advisory Committee(CRDAC)が開催され,ESAの安全性に 関して詳細な検討がされたが,米国における添付文書の更なる変更はなされていない。
欧州においては,2007年4月に欧州医薬品委員会(CHMP)が,ESA の安全性について検討 を始め,ESAによる目標Hb濃度を10~12 g/dLに統一し,「Hb濃度が12 g/dLを超えてはなら ない」と添付文書が改訂された。2009年には ERBPとして KDOQIと同様に「Hb 濃度13 g/dL を超えるべきではない」とされた。
これらの海外の状況を踏まえ,国内ガイドラインの目標 Hb 濃度の上限あるいは ESA の減 量・休薬基準について慎重に検討がなされた。CHOIR 試験に参加した約1/3の患者は,心筋梗 塞,脳卒中の既往を有する,又は冠動脈バイパス術や経皮的冠動脈インターベンション後,四 肢切断後の患者であり,重篤な CVD を有する患者を多く含む集団であった。しかし,国内の
rHuEPO 製剤の大規模前向き観察研究の中間報告29)による血液透析導入患者の合併症及び治療
歴と比較すると,CHOIR試験では CVDの頻度及び重篤度ともに極めて高く,国内の平均的な 保存期患者の背景とは大きく異なっている7)。
また,一連のガイドラインや添付文書の改訂を促すこととなった CHOIR 試験において,追 加解析の結果が2008年8月に報告された30)。高 Hb 濃度群に割り付けられた患者の中でも到達 した Hb 濃度が高い患者のほうがむしろ予後が良いこと,1回最高投与量が高い群(20,000 IU 以上)の方が低い群(20,000 IU 未満)に比べ予後が悪いこと,これらの因子で調整した Cox ハザードモデル解析では,割り付けられた目標 Hb 濃度の群と予後の関連性はなくなり,特に 高投与量の使用が予後悪化との関連性を最も説明できる因子であった。このことから,目標 Hb濃度が高いことと予後悪化の関連性は確認できなかったと報告されている。
重篤な CVD を有し,Hb 濃度の上昇に多量の ESA を必要とする患者,あるいは何らかの要
因でrHuEPO製剤に対する不応性が出現するような病態では予後が不良となることは当然の結
果と考えられ,これまでの研究報告を総合的に判断し,国内すべての保存期及び腹膜透析患者 に対して目標Hb濃度の上限を12 g/dLに制限する根拠は薄いと結論された。そこで,保存期及 び腹膜透析患者においては,目標Hb濃度の上限は設定されず,13 g/dLを超える場合に減量・
休薬することが推奨された。ただし,重篤な心・血管系疾患の既往や合併のある患者,あるい は医学的に必要のある患者には12 g/dLを超える場合に減量・休薬することが推奨された。
その後,海外では2009年11月に,Ⅱ型糖尿病を有する CKD 患者を対象として,ダルベポエ チン アルファを投与して目標 Hb 濃度を13 g/dL とした群(DA 群)とプラセボを投与した群
( プ ラ セ ボ 群 ) の2群 を 比 較 し た 二 重 盲 検 比 較 試 験 で あ る TREAT(The Trial to Reduce Cardiovascular Events with Aranesp Therapy)試験55)の結果が報告された。主要評価項目である 死亡及び心・血管系疾患の複合イベントの発現率は,DA 群とプラセボ群で有意差は認められ ず,脳卒中の発現率は DA群5.0%に対してプラセボ群2.6%と,DA群で有意に高い結果が示さ れた。また,悪性腫瘍の既往のある患者では,がんによる死亡率が DA 群で有意に高かった
(Log rank検定:P = 0.002)。このことから,日本腎臓学会は,CKD患者におけるESAの安 全性を検証すべく,国内調査研究であるSurveillance of Epoetin-Adverse Events of Stroke and Cancer
(SEASCAN)56)を緊急で実施した。その結果,rHuEPO 製剤非投与群,短期投与群及び長期投 与群の3群間で,症候性脳梗塞の発現率に有意差はなく,がんの新規診断・増悪においても3群 間に有意差は認められないことが2010年6月に報告された。
(3) ESAの投与量及びHb濃度の是正速度
Hb濃度の是正速度について,rHuEPO製剤の臨床使用の開始当初は,高血圧や血圧上昇の懸 念から週あたりの貧血改善速度が0.3~0.4 g/dLを超えないことが安全性の観点から重要と考え られていた。近年,ESA による血圧上昇は血液粘度の上昇などに伴う随伴症状との理解が定 着し,早期より高血圧を予防する手段が用いられ,2000年以降は急激な血圧上昇に伴う高血圧 性脳症などの合併症はほとんど報告されなくなっており,週あたり Hb 濃度0.5 g/dL 以内の上 昇速度であれば問題ないと考えられている7)。更に,投与開始時の Hb 濃度が低ければ,可能 な限り輸血を避けるべく,より急速な Hb 濃度の改善が求められる場合があることも指摘され ている。また,ESA の投与に際しては,患者の病期に応じて是正目標値や是正速度を設定し,
投与量を決定するべきであることがガイドラインに示されている。
2.5.1.2 国内及び海外における開発の経緯 2.5.1.2.1 国内における開発の経緯
本剤の開発の経緯を試験ごと及び対象ごとに図 2.5.1.2.1-1に示した。
第Ⅰ相試験(JP16690:単回静脈内投与)は,2002年4月より日本人及び白人の健康成人男性 を対象に米国( )で実施し,本剤に対する忍容性,薬物動態及び薬力学に関して,人種 間に大きな違いがないことを確認した。引き続いて,第Ⅰ相試験(JP17138:単回皮下投与試 験)を2003年9月より健康成人男性を対象に国内で実施した。反復投与試験については,健康 成人を対象とした場合,必要以上の造血が懸念されたことから,腎性貧血患者を対象に実施す ることとした。
そこで,2004年7月よ り腎性貧血患者を対象に前期第Ⅱ相試験(JH18120:血 液透析,
JH18084:保存期)の2試験を実施し,反復静脈内投与による有効性,安全性及び薬物動態を 検討した。また,腎性貧血患者を対象とした臨床薬理試験として,2004年7月より単回静脈内 投与試験(JP18117:血液透析,JP18118:保存期)の2試験,2005年10月より単回皮下投与試 験(JP19454:保存期,JP19455:腹膜透析)の2試験を実施した。
血液透析患者では,JH18120,JP18117の試験成績を基に2005年10月より用量設定を目的に後 期第Ⅱ相試験として二重盲検比較試験(JH19307:静脈内投与),rHuEPO 製剤からの切替維 持試験(JH19308:静脈内投与)の2試験を実施した。また,保存期患者における静脈内投与 での後期第Ⅱ相試験は,血液透析と同様に JH18084,JP18118の試験成績を基に用量設定試験
(JH19400)として実施した。保存期患者における皮下投与での後期第Ⅱ相試験は,第Ⅰ相単 回 皮 下 投 与 試 験 終 了 後 , 海 外 試 験 成 績 を 参 考 に2005年4月 よ り 用 量 設 定 試 験
(JH18512/JH18537*)として実施した。
第Ⅱ相試験成績が得られた時点で, 及び について,医
薬品第Ⅱ相試験終了後相談を20 年 月 日に実施した。本相談では, ,
,
,また,
助言されたことから,20 年 月 日, 月 日に再度医薬品追 加相談を実施した。
これらの相談における機構の助言に基づき,第Ⅲ相試験として血液透析患者を対象とした貧 血改善・改善維持試験(JH20562:静脈内投与),切替維持試験(JH20563:静脈内投与),
エポエチン ベータとの二重盲検比較試験(JH20876:静脈内投与),保存期患者を対象とした 切 替 維 持 試 験 (JH20566: 静 脈 内/皮 下 投 与 ) , エ ポ エ チ ン ベ ー タ と の 非 盲 検 比 較 試 験
(JH20565:皮下投与),腹膜透析患者を対象とした切替維持試験(JH20564:静脈内/皮下投 与)の6試験を2007年1月より順次実施し,2009年7月に承認申請を行った。
第Ⅲ相試験では,JH20565の主要評価項目である「Hb 濃度維持率」の非劣性が示されなかっ たため,20 年 月 日の初回面談及びその後20 年 月 日, 月 日, 月 日及び 月 日の 回に亘り機構との協議の場がもたれた。その中で,申請者は JH20565が比較試験である ため,他の臨床試験や実地医療とは異なり医師の裁量による用量調整を厳格に制限したことが 影響したものの,「Hb 濃度維持率」と同様に適切な評価指標である「平均 Hb 濃度」では,
rHuEPO 製剤と同等の有効性が示されたこと,また,医師の裁量により適切に用量調整された
他の臨床試験における「Hb 濃度維持率」は,JH20565のエポエチン ベータ群に比べて劣るも のではないことを説明した。これらのことから,本剤は適切な用量調整を行うことにより目標 とするHb濃度の範囲に維持できるESAであり,本剤の有効性及び安全性は用量調整方法を含 めて提出した申請資料から確認できると考えた。しかしながら,機構は,
,
, との見解を示した。
以上の経緯を踏まえた上で,申請者は用量調整方法に関して申請データパッケージを補完す
るための無作為化非盲検比較試験(JH22757:皮下投与)を計画し,20 年 月 日に本試験 の試験デザインや評価方法等を定めた試験計画の骨子について機構と合意した上で,2010年1 月から試験を開始するに至った。なお,JH22757は2008年に日本透析医学会から「慢性腎臓病 患者における腎性貧血治療のガイドライン」7)が公表されたことから,本ガイドラインに基づ いて対象患者及びHb濃度の目標値を設定した。
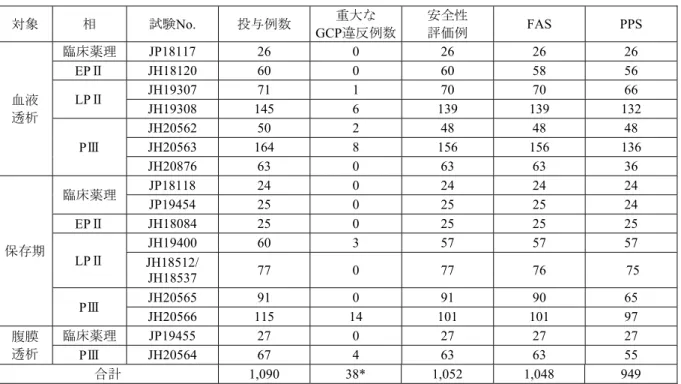
以上,健康成人を対象とした第Ⅰ相試験2試験,血液透析患者を対象とした7試験,保存期患 者を対象とした8試験,腹膜透析患者を対象とした2試験の計19試験を臨床データパッケージと した。
*:JH18537は JH18512の 継 続 試 験 で あ る た め1試 験 と し て 集 計 し , 図 表 下 に 脚 注 で
「*:JH18512+JH18537」と示した。
図 2.5.1.2.1-1 開発の経緯
試験No. 対象 投与
経路 2002年 2003年 2004年 2005年 2006年 2007年 2008年 2009年 2010年
JP16690
健康成人
i.v. ④ ⑫ PⅠ
JP17138 s.c. ⑨ ⑥PⅠ
JP18117
血液透析 i.v.
⑦ ⑤臨床薬理
JH18120 ⑦ ⑤EPⅡ
JH19307 ⑩ ⑥LPⅡ
JH19308 ⑩ ②LPⅡ
JH20562 ① ③PⅢ
JH20563 ① ④PⅢ
JH20876 ⑤ ③PⅢ
JP18118
保存期 i.v.
⑨ ⑩ 臨 床薬理
JH18084 ⑦ ⑤EPⅡ
JH19400 ⑩ ④LPⅡ
JP19454
s.c.
⑪ ⑨ 臨床 薬理
JH18512/JH18537 ④ ⑧LPⅡ
JH20565 ⑤ ⑦PⅢ
JH20566 i.v.
s.c. ① ⑦PⅢ
JH22757 s.c. ① ⑧PⅢ
JP19455
腹膜透析
s.c. ⑩ ④臨床薬理
JH20564 i.v.
s.c. ① ⑤PⅢ
各試験の左の丸数字は第1症例の同意取得月,右の丸数字は最終症例の最終検査月を示す。
CERA 2.5 臨床に関する概括評価16Page
2.5.1.2.1.1 臨床データパッケージ
本申請で評価資料とした本剤の19臨床試験の一覧を表 2.5.1.2.1.1-1に示した。
表 2.5.1.2.1.1-1 臨床試験一覧
相 対象 投与 経路
試験名
(試験 No.) 用法・用量 試験デザイン
投与期間(週) 投与例数1)
実施期間2) 上:開始年月 下:終了年月
PⅠ 健康 成人
i.v. 単回投与試験
(JP16690) プラセボ, 0.8, 1.6, 3.2 μg/kg
無作為化単盲検 プラセボ対照 比較試験,単回
日本人: 36 白人: 36
2002年4月 2002年12月
s.c. 単回投与試験
(JP17138) プラセボ, 0.4, 0.8, 1.6, 3.2 μg/kg
無作為化二重盲検 プラセボ対照 比較試験,単回
40 2003年9月 2004年6月
臨床 薬理
血液
透析 i.v. 単回投与試験
(JP18117) 100, 150, 200 μg 無作為化非盲検
群間比較試験,単回 26 2004年7月 2005年5月
保存期
i.v. 単回投与試験
(JP18118) 100, 150, 200 μg 無作為化非盲検
群間比較試験,単回 24 2004年9月 2005年10月 s.c. 単回投与試験
(JP19454) 100, 200, 300 μg 無作為化非盲検
群間比較試験,単回 25 2005年11月 2006年9月 腹膜
透析 s.c. 単回投与試験
(JP19455) 100, 200, 300 μg 無作為化非盲検
群間比較試験,単回 27 2005年10月 2006年4月
EPⅡ 血液
透析 i.v. 貧血改善試験
(JH18120) 初期投与期:12.5, 25, 50, 75 μg/2週 無作為化非盲検
群間比較試験,6 60 2004年7月 2005年5月 保存期 i.v. 貧血改善試験
(JH18084) 初期投与期:12.5, 25, 50 μg/2週 無作為化非盲検
群間比較試験,6 25 2004年7月 2005年5月
LPⅡ 血液 透析 i.v.
貧血改善試験
(JH19307) 初期投与期:25, 50, 75 μg/2週 無作為化二重盲検
群間比較試験,16 70 2005年10月 2006年6月 切替維持試験
(JH19308)
切 替 投 与 期 (8週 ) :50, 100, 150, 200 μg/4週 維持投与期:25~300 μg/4週
無作為化非盲検 群間比較試験
48
139 2005年10月 2007年2月
保存期 i.v.
貧血改善・
改善維持試験
(JH19400)
初期投与期:25, 50, 75 μg/2週 維持投与期:25~300 μg/4週
無作為化非盲検 群間比較試験
48~50
57 2005年10月
2007年4月
s.c.
貧血改善・
改善維持試験
(JH18512/
JH18537)
初期投与期:25, 50, 75 μg/2週 維持投与期:12~300 μg/4週
無作為化非盲検 群間比較試験
48~50
JH18512:
77 JH18537:
50
2005年4月 2006年8月 1)重大なGCP違反例を除いた投与例数
2)開始年月は第1症例の同意取得月,終了年月は最終症例の最終検査月
3)16週間の維持投与期と24週間の継続投与期
相 対象 投与 経路
試験名
(試験 No.) 用法・用量 試験デザイン
投与期間(週) 投与例数1)
実施期間2) 上:開始年月 下:終了年月
PⅢ 血液 透析
i.v.
貧血改善・
改善維持試験
(JH20562)
初期投与期:50 μg/2週 維持投与期:25~300 μg/4週
非盲検一般臨床試験
24~26 48 2007年1月
2008年3月 i.v. 切替維持試験
(JH20563)
切替投与期(8週):100, 150 μg/4週 維持/継続投与期3):25~400 μg/4週
非盲検一般臨床試験
48 156 2007年1月
2008年4月
i.v. 切替維持試験
(JH20876)
切替投与期(8週):
Ro50-3821:100, 150 μg/4週
エポエチン ベータ:2250, 3000, 4500, 6000 IU/週
維持投与期:
Ro50-3821:50~250 μg/4週 エポエチン ベータ:750~9000 IU/週
無作為化二重盲検 比較対照試験
24
Ro50-3821:
63 エポエチン ベータ:
63
2007年5月 2008年3月
保存期 s.c.
貧血改善・
改善維持試験
(JH20565)
初期投与期:
Ro50-3821:25 μg/2週 エポエチン ベータ:6000 IU/週 維持投与期:
Ro50-3821:25~250 μg/4週 エポエチン ベータ:3000~12000 IU/2週
無作為化非盲検 比較対照試験
24~26
Ro50-3821:
91 エポエチン ベータ:
89
2007年5月 2008年7月
i.v.
s.c.
切替維持試験
(JH20566)
切替投与期(8週):100, 150 μg/4週 維持/継続投与期3):25~400 μg/4週
非盲検一般臨床試験 48
i.v.: 31 s.c.: 70
2007年1月 2008年7月
s.c.
貧血改善・
改善維持試験
(JH22757)
初期投与期:
Ro50-3821:25 μg/2週 エポエチン ベータ:
6000 IU/週又は6000 IU/2週 維持投与期:
Ro50-3821:25~250 μg/4週 エポエチン ベータ:1500~12000 IU/2週
無作為化非盲検 比較対照試験
24~26
Ro50-3821:
45 エポエチン ベータ:
43
2010年1月 2010年8月
腹膜 透析
i.v.
s.c.
切替維持試験
(JH20564)
切替投与期(8週):100, 150 μg/4週 維持/継続投与期3):25~400 μg/4週
非盲検一般臨床試験 48
i.v.: 28 s.c.: 35
2007年1月 2008年5月 1)重大なGCP違反例を除いた投与例数
2)開始年月は第1症例の同意取得月,終了年月は最終症例の最終検査月
3)16週間の維持投与期と24週間の継続投与期
(1) 第Ⅰ相試験
健康成人男性を対象とした第Ⅰ相試験として,本剤単回投与後の薬物動態を検討する目的で,
日本人及び白人での単回静脈内投与試験(JP16690:プラセボ,0.8,1.6,3.2 μg/kg),日本人 での単回皮下投与試験(JP17138:プラセボ,0.4,0.8,1.6,3.2 μg/kg)を実施した。その結 果,静脈内及び皮下投与ともに,AUC は3.2 μg/kg まで用量に比例して増加し,消失半減期
(t1/2)はそれぞれ66.2~75.8時間及び96.2~123時間であり,rHuEPO製剤に比較してt1/2の延長 が 確 認 さ れ た 。 安 全 性 に も 大 き な 問 題 は な か っ た 。 ま た , 日 本 人 と 白 人 の 薬 物 動 態
(Pharmacokinetics:PK)プロファイルに大きな違いはなかったため,海外の臨床試験成績を 参考に,反復投与後の PK 及び安全性の検討は腎性貧血患者を対象とした前期第Ⅱ相試験で検 討することとした。
(2) 臨床薬理試験
血液透析及び保存期患者を対象に,本剤単回静脈内投与後の薬物動態を検討する目的で臨床 薬理試験を実施した(JP18117,JP18118:100,150,200 μg)。また,同様に保存期及び腹膜 透析患者を対象に,本剤単回皮下投与後の臨床薬理試験を実施した(JP19454,JP19455:100,
200,300 μg)。その結果,静脈内及び皮下投与のいずれの投与経路においても t1/2は rHuEPO
製剤に比較して大幅に延長しており,AUC は用量に比例して増加した。また,透析期(血液 透析,腹膜透析)及び保存期の病期間での PKプロファイルは類似していた。安全性について 問題は認められなかった。
(3) 前期第Ⅱ相試験
前期第Ⅱ相試験として,本剤の反復投与後の薬物動態,有効性及び安全性を検討する目的で,
血液透析患者を対象とした試験(JH18120:静脈内投与,2週に1回12.5,25,50,75 μg),保 存期患者を対象とした試験(JH18084:静脈内投与,2週に1回12.5,25,50 μg)の2試験を実 施した。その結果,t1/2の延長が認められ AUC は用量に比例して増加した。また,3回反復投 与後の薬物動態は初回投与時と同様であり,血液透析及び保存期患者の PK プロファイルも同 様であることが示された。Hb 濃度は用量に依存して増加し,貧血改善効果が認められた。安 全性について問題は認められなかった。
(4) 後期第Ⅱ相試験
後期第Ⅱ相試験として,血液透析患者では,初期投与期における至適用量の検討を目的とし た貧血改善試験(JH19307),及び,rHuEPO 製剤から本剤への切替初回用量の検討を目的と した切替維持試験(JH19308)を実施した。また,保存期患者では,初期投与期における至適 用 量 の 検 討 を 目 的 と し た 貧 血 改 善 ・ 改 善 維 持 試 験 (JH19400: 静 脈 内 投 与 , JH18512/JH18537:皮下投与)を実施した。なお,JH19308,JH19400,JH18512/JH18537の3試 験ではそれぞれ切替及び初期投与期終了後に維持投与期に移行し,長期投与時の有効性及び安 全性を検討した。
JH19307(2週に1回25,50,75 μg)においては,rHuEPO製剤休薬後Hb濃度が9.5 g/dL未満 の血液透析患者を対象に二重盲検法により至適用量の検討を行った結果,50 μg 以上で高い貧 血改善効果が得られ,安全性にも問題がなかったことから,血液透析患者における初期用量は
1回50 μgが妥当であると判断された。
JH19308においては,rHuEPO製剤の投与により Hb濃度が安定している患者を対象に,開始
前の rHuEPO 製剤の投与量が週あたり4500 IU 未満(4500 IU 未満群)の患者には50,100,
150 μg,週あたり4500 IU以上(4500 IU以上群)の患者には100,150,200 μgのいずれかを4
週に1回,8週間静脈内投与した。その結果,主要評価項目である Hb 濃度の回帰直線の傾きか ら,rHuEPO製剤からの切替初回用量は4500 IU未満群では100 μg,4500 IU以上群では150 μg が至適であると考えられた。
JH19400(2週に1回25,50,75 μg)及びJH18512/JH18537(2週に1回25,50,75 μg)におい ては,投与開始前4週間以内に rHuEPO製剤が投与されていない Hb濃度10.0 g/dL未満の保存 期患者を対象に至適用量の検討を行った。その結果,静脈内及び皮下投与経路ともに十分な貧 血改善効果を得るためには50 μg 以上が必要と考えられたが,医薬品第Ⅱ相試験終了後相談で の有効性及び安全性に関する助言を総合的に勘案して,第Ⅲ相試験での初期用量は25 μg とす ることとした。
JH19308においては,8週間の切替投与期終了後,Hb濃度を10.0~12.0 g/dLに維持するよう4
週に1回の頻度で用量を適宜増減し合計48週間の維持投与を行った。また,JH19400及び JH18512/JH18537では,初期投与期終了後,投与頻度を2週に1回から4週に1回に切り替え,Hb 濃度を11.0~13.0 g/dLに維持するよう用量を適宜増減し合計48~50週間の維持投与を行った。
その結果,血液透析及び保存期患者ともに用量を適宜増減することにより Hb 濃度を目標値内 に維持することが可能であった。
(5) 第Ⅲ相試験
第Ⅲ相試験として,血液透析患者では貧血改善・改善維持試験(JH20562),切替維持試験
(JH20563),エポエチン ベータとの二重盲検比較試験(JH20876)の3試験,保存期患者では 切 替 維 持 試 験 (JH20566: 静 脈 内/皮 下 投 与 ) , エ ポ エ チ ン ベ ー タ と の 非 盲 検 比 較 試 験
(JH20565:皮下投与),エポエチン ベータとの非盲検比較試験(JH22757:皮下投与)の3試 験,腹膜透析患者では切替維持試験(JH20564:静脈内/皮下投与)の1試験,計7試験を実施し た。
JH20562(血液透析)においては,血液透析導入後 rHuEPO 製剤の投与経験がない患者を対
象に本剤を2週に1回50 μg 投与した結果,高い貧血改善効果が得られ安全性にも問題はなかっ たことから,血液透析患者における初期用量は1回50 μg が妥当であることが確認された。
JH20563(血液透析),JH20566(保存期),JH20564(腹膜透析)の切替維持試験においては,
いずれの試験でも本剤を4週に1回適宜増減することにより,Hb 濃度が長期に亘り目標値内に 維持され,安全性に関しても問題はなかった。JH20876(血液透析)では評価期間における平 均 Hb濃度変化量の差(95%信頼区間:95%CI)は0.457 g/dL(0.166~0.747 g/dL)と同等性マ ージンの ± 1.0 g/dLの範囲であり,エポエチン ベータに対する同等性が検証された。安全性に ついては,有害事象の発現率とその内容に両群で違いは認められなかった。また,JH20565
(保存期)では主要評価項目のHb濃度維持率においてエポエチン ベータとの非劣性が示され なかったが,副次評価項目の評価期間における両群の平均Hb濃度の差(95%CI)は0.532 g/dL
(0.283~0.782 g/dL)であり, ± 1.0 g/dL の範囲内であった。Ro50-3821群の平均 Hb 濃度は
11.2~11.9 g/dLの間を推移していた。有害事象の発現率とその内容に両群で違いは認められな
かった。
JH22757(保存期)における主要評価項目は「①Ro50-3821群での目標Hb濃度12.0 g/dLと評
価期間における平均 Hb 濃度との差」及び「②評価期間における平均 Hb 濃度の Ro50-3821群 とエポエチン ベータ群との差」とし,①については,目標値と平均 Hb濃度の差の95%信頼区 間が ± 1.0 g/dLの範囲内であることを検証した。②については,①が検証された場合に本評価 を行い,エポエチン ベータ群に対する平均値の差の95%信頼区間の下限が − 0.75 g/dL 以上で ある場合,エポエチン ベータ群に対して非劣性が検証されると定義した。その結果,主要評 価項目①である本剤群の目標 Hb 濃度12.0 g/dL と評価期間における平均 Hb 濃度との差
(95%CI)は, – 0.44 g/dL( − 0.65~ − 0.23 g/dL)と ± 1.0 g/dLの範囲内にあったことから,
本剤の投与により目標とする Hb 濃度に維持されることが検証された。また,主要評価項目② であるエポエチン ベータ群に対する本剤群の評価期間における平均 Hb 濃度の差の95%CI は
0.17~0.78 g/dLと非劣性マージン − 0.75 g/dL以上を満たし,エポエチン ベータに対する貧血
改善維持効果の非劣性が検証された。安全性については,有害事象の発現率とその内容に両群 で違いは認められなかった。




![図 2.5.3.4.1-1 静脈内及び皮下投与後の血清中 Ro50-3821濃度の推移(平均値) [5.3.3.5-1 図2-17 再掲] 図 2.5.3.4.1-2 静脈内及び皮下投与後のトラフ値の推移(平均値 ± SD) [5.3.5.1-3 第2版 図11.4.6.2-1,5.3.5.1-7 第2版 図11.4.7.2-1,5.3.5.1-5 図11.4.7.2-1 改変] 2.5.3.4.2 投与経路間の薬力学的反応の比較 投与経路間の薬力学的反応の比較は,同一試験内で本剤100,1](https://thumb-ap.123doks.com/thumbv2/123deta/7608109.2540736/41.892.124.768.111.736/静脈内及皮下投与血清濃度推移トラフ経路間薬力学経路間薬力学.webp)
![表 2.5.3.7-1 PK/PD モデルにより算出された Hb 濃度維持率 評価期間の平均 Hb 濃度 患者の割合(%) 95%CI 12.0 g/dL 超 33.8 30.9~36.7 10.0~12.0 g/dL 64.7 61.7~67.7 10.0 g/dL 未満 1.5 0.7~2.3 [5.3.3.5-2 表2.3-2 改変] 表 2.5.3.7-2 用量調整基準 投与期 項目 JH20565 条件1 条件2 初期 投与期 初期用量 25 μg 開](https://thumb-ap.123doks.com/thumbv2/123deta/7608109.2540736/45.892.117.787.54.977/PKPDモデルにより算出濃度維持評価期間平均投与期投与期初期用量.webp)

