PK 解析の基礎
Phase I を中心に
Introduction to Pharmacokinetic Analysis
—Focus on Phase I Study—
猪川和朗∗1・ 田中 潤∗2
Kazuro Ikawa∗1 and Jun Tanaka∗2
∗1広島大学大学院臨床薬物治療学
∗2中外製薬株式会社
∗1Department of Clinical Pharmacotherapy, Hiroshima University ∗2
Chugai Pharmaceutical Co., Ltd. e-mail:[email protected]
We describe fundamental knowledge of pharmacokinetics analysis for phase I trials, particularly focusing on basic parameters (such as bioavailability, volume of distribu-tion, fraction unbound, clearance), estimation and analysis methods (such as com-partmental and non-comcom-partmental), points to consider (such as steady state and dose proportionality). The NCA is an abbreviation for Non Compartmental Anal-ysis, and the meaning is pharmacokinetic analysis without pharmacokinetic model. There is something that we should consider in NCA such as AUC calculation method, handling method of not detectable concentrations, point selection for λzcalculation,
and selection of sampling time. Steady state occurs when the overall intake of a drug is equilibrium with its elimination. At steady state the mean plasma concentrations of the drug are similar by any dosing interval. In practice, it is generally considered that steady state is reached when a time of 5 times the half-life for a drug. For the dose proportionality, the measures of exposure, such as maximal blood concentration (Cmax), area under the curve from 0 to infinity (AUC), are proportional to the dose. The three methods, Analysis of variance of the PK response, normalized by dose, linear regression and power model, are used to assess dose proportionality.
Key words: volume of distribution, clearance, compartmental analysis, non com-partmental analysis, steady state, dose proportionality.
1. Pharmacokinetics(PK)総論
Pharmacokinetics(PK)とは一般に,薬物(用法用量)が投与された後の,生体内で吸収,分布, 代謝,排泄という一連の過程を経た,体内薬物濃度(量)の推移をいう.通常,「薬物用法用量 −
薬物が投与された後に体内では,吸収(absorption),分布(distribution),代謝(metabolism), 排泄(excretion)の過程が並行して進行するため,PK を完全に解明することは現実的に不可能で ある.しかし,これらの過程を分解して,記述・説明しようとするための基本的なパラメータが 確立されてきた.それが,5 つの基本的な PK パラメータ,⃝1 バイオアベイラビリティ,⃝2 分布 容積,⃝3 血中非結合形分率,⃝4 クリアランス,⃝5 尿中排泄率である.以下,順次説明する. 2. PK パラメータ ⃝1 バイオアベイラビリティ(bioavailability: F )とは,血管外(経口など)で投与された薬物量(D) と全身循環血中に到達した薬物量を関連づけるパラメータであり,0 − 1 の値をとる.全身循環血 中に到達した薬物量は F · D で表される. ⃝2 分布容積(volume of distribution: Vd または V )とは,体内薬物量と体内薬物濃度を関連づけ る “仮想” の体液量である.投与薬物量は既知だが,投与後の体内薬物量(A)は未知であり,観測 できるのは体内薬物濃度(C)だけであるため,このパラメータの存在意義がある.体内薬物量は, A = Vd · C で表される.Vd の値が小さい(特に 3 L 以下の)場合,その薬物は血管内に留まると 解釈され,Vd が大きい(特に 36 L 以上の)場合には,薬物は血管外の体液・組織にも広く分布す ると解釈される.図 1 に示すとおり,分布容積の概念には V1(1 番目,通常は中心コンパートメ ントの分布容積),Vdβ(ベータ相,すなわち 2 番目の消失相における分布容積),Vdss(定常状 態における分布容積)など各種あり,データの内容や解析の目的に応じて使い分けている.
⃝3 血中非結合形分率(fraction unbound in blood: fuB)は,fuB · 血中総濃度として,血中遊離
形濃度を規定するパラメータであり,通常,薬物の血漿タンパク(アルブミンや α1 酸性糖タンパ ク)への結合率を 1 から引いた値である.血漿タンパクに結合していない遊離形の薬物が標的部位 に到達し,薬理作用を発現するため,本来は血中非結合形濃度が重要となる.しかし,薬物血中 濃度はほとんど総濃度(結合形濃度 + 非結合形濃度)として測定されているため,血中非結合形分 率が必要となる. ⃝4 クリアランス(clearance: CL)は,処理臓器による血液清浄速度と捉えられる(図 2).薬物は, 血流によって動脈血から静脈血へ移動して処理臓器(肝臓または腎臓)を通過中に消失すると考え 図 1. 分布容積(V1, Vdβ, Vdss)
図 2. クリアランス
られるため,CL の単位 [L/h] は,血流速度(Q)の単位 [L/h] に等しい.生理学的には,動脈血中 薬物濃度 Ca,静脈血中薬物濃度 Cv,抽出比 E = (Ca − Cv)/Ca として,CL[L/h] = Q[L/h] · E で表される.
⃝5 尿中排泄率(cumulative amount of drug excreted in urine: Ae)とは,投与薬物量のうち,肝
臓で代謝されず “未変化体” として腎臓で排泄された体外薬物量の比率である.なお,代謝物とし て腎排泄された薬物は含まないので,注意が必要である.Ae = 累積尿中未変化体排泄量/投与薬 物量として,体全体の血液清浄速度のうちの腎臓が担う比率(腎クリアランスの比率)を示す.例 えば Ae = 0.7 ならば,体全体の血液清浄速度の 70 %を腎臓が担うと解釈し,全身クリアランス = 腎クリアランス(70 %)+ 肝クリアランス(30 %)と解釈する. すべての薬物はそれぞれ⃝1 ⃝5 について固有の値を有するため(ただし⃝1 は血管外投与薬物の み),各値での分類に基づいて薬物が特徴づけされることになる. これらの基本的 PK パラメータに加えて,よく用いられる PK パラメータとしては,薬物濃度 − 時間曲線下面積(area under drug concentration-time curve: AUC),平均滞留時間(mean
resi-dence time: MRT),最高薬物濃度(Cmax),最高薬物濃度到達時間(tmax),最低薬物濃度(Cmin),
消失半減期(t1/2)などがある. 3. PK パラメータ推定方法 PK パラメータの推定方法は,図 3 に示すとおり,特定の数理モデルに依存しないモーメント解 析(通常,ノンコンパートメント解析と呼称される)と特定の数理モデルに基づいたコンパートメ ントモデル解析に大別される.ノンコンパートメント解析は,簡便な推定計算で PK を評価でき るという利点がある.コンパートメントモデル解析では,モデルを構築した後にシミュレーショ ンを行うことで,別の特定条件下での PK も予測できるという利点がある. この点に関しては,厚生労働省通知『医薬品の臨床薬物動態試験について』(平成 13 年 6 月 1 日, 医薬審発第 796 号)の「7.1. 薬物動態解析法」に具体的な記載がある.「標準的な薬物動態試験法で は,十分な測定時点数を確保し,モデルに依存しない解析法により,血中濃度 − 時間曲線下面積
(AUC),クリアランス,最高血中濃度(Cmax),最低血中濃度(Cmin),最高血中濃度到達時間
図 3. PK パラメータの推定手法 図 4. モーメント(ノンコンパートメント)解析 ンパートメントモデル等に基づくモデル解析を利用すると,上記薬物動態パラメータに加え,速 度定数,分布容積(V1, Vdβ, Vdss)に関する情報が得られる.観測された血中濃度 − 時間推移を 記述しうるモデルを構築することは,用量や投与法の違いによる血中濃度の変化を予測し,併せ て個別投与設計に活用する上で,また,PK/PD 解析へ発展させる上で有意義である」. 図 4 に示すとおり,ノンコンパートメント解析では,単純に台形の面積を足し合わせて AUC や MRT などの値を求める.ただし,観測点に影響を受けるため,注意が必要である. 一方,コンパートメント解析(図 5,図 6)では,体内に特定の区画を想定し,質量保存の法則 に基づいた物質収支式が成立するとして,PK パラメータを求めていく.この物質収支式は,薬 物移動速度として捕らえるため微分方程式となるが,薬物濃度(C)ではなく,薬物量(X)を基準 とした式である点に注意が必要である.コンパートメント解析からのクリアランス定義式は, dX/dt [mg/h] = −CL [L/h] · C [mg/L] で表され(図 5),t1/2の算出式は図 6 に示されるとおりで ある. Phase I を含め医薬品開発における PK 解析で繁用されるコンパートメントモデルは,点滴静 注 1 コンパートメントモデル(図 7),経口投与 1 コンパートメントモデル(図 8),点滴静注 2 コ ンパートメントモデル(図 9),経口投与 2 コンパートメントモデル(図 10)の 4 つである. コンパートメントモデルでの物質収支式(微分方程式)において,投与経路が経口投与,筋肉注 射,皮下注射などは,体内流入速度が薬物量の 1 乗に比例する様式に従うと仮定する(例えば図 8 の吸収速度定数 Ka).投与経路が点滴静注などでは,体内流入速度が薬物量の 0 乗に比例,すな わち薬物量にかかわらず一定になると仮定する(例えば図 7 の点滴速度 R).また,薬物が対象コ
図 5. コンパートメントモデル(速度論)解析 図 6. 1-compartment model(bolus 静注)での消失半減期 図 7. 点滴静注 1 コンパートメント PK モデル ンパートメントへ流入する場合はプラス符号を,薬物が対象コンパートメントから流出する場合 にはマイナス符号を付け,コンパートメントの数に応じた連立方程式が成立する(図 10 の経口投 与 2 コンパートメントモデルでは,コンパートメントの数は 3 つで,式の数も 3 つとなる). コンパートメントモデル解析の手順に関しては,⃝1 モデル構築(データの視覚化,コンパートメ ント数・パラメータ数の決定などを含む),⃝2 モデルパラメータの推定(推定アルゴリズムや解析 プログラムなどの決定を含む),⃝3 パラメータ推定値の評価(信頼区間,予測精度などを含む)の流 れで進み,必要に応じて⃝4 特定条件下でのシミュレーションが実施される.前述の通知『医薬品 の臨床薬物動態試験について』の「7.2. 統計解析方法」には「これら解析の根拠とした薬物動態 モデル,薬物動態パラメータなどの推定方法,解析に用いたソフトウェア(パッケージ),はずれ
図 8. 経口投与 1 コンパートメント PK モデル
図 9. 点滴静注 2 コンパートメント PK モデル
図 10. 経口投与 2 コンパートメント PK モデル
値や定量下限未満の濃度データの取り扱いについて明記する」と記載されている.
4. NCA(Non Compartmental Analysis) 4.1 NCA
NCA とは Non Compartmental Analysis の略語であり,薬物動態モデルに依存しない解析方法 を意味し,Compartmental Analysis,すなわち薬物動態モデルを仮定した解析方法の対義語とな
る.薬物動態解析において NCA を用いる主な理由としては,薬物動態モデルを仮定しないため, 真の薬物動態モデルがどの様なモデルであっても問題ない,モデルを選択する際のバイアスが入 らない,各薬物動態パラメータを求めるための適切な採血時点を設定できれば正確な薬物動態パ ラメータを求めることができる,といった点が挙げられる.その一方で,採血時点が適切でない 場合には,求まった薬物動態パラメータの信頼性が低下する事となる. NCA において主に対象となるのは血漿中濃度及び尿中濃度である.血漿中濃度の NCA で求め るパラメータは「医薬品の臨床薬物動態試験について」(厚生労働省, 2001)では以下のように定め られている.「標準的な薬物動態試験法では,十分な測定時点数を確保し,モデルに依存しない解 析法により,血中濃度 − 時間曲線下面積(AUC),クリアランス,最高血中濃度(Cmax),最低血 中濃度(Cmin),最高血中濃度到達時間(tmax),定常状態分布容積(Vdss),平均滞留時間(MRT), 半減期(t1/2)等を求める」.また,尿中濃度の NCA で求めるパラメータは「後発医薬品の生物学 的同等性試験ガイドライン」(厚生労働省, 2012)では以下のように定められている.「尿を採取体
液とする場合は,Aet, Aeτ, Ae∞, Umax及び Uτを AUCt, AUCτ, AUC∞, Cmax及び Cτに代わ
るパラメータとして用いる」. 臨床試験において NCA を行う場合にプロトコール及び解析計画書に記載が必要となる事項の例 を以下に示す.AUC や見かけの消失速度定数(以下 λz)にはいくつかの計算方法があるため,ど のような方法を用いるかを事前に明記しておく必要がある. 【プロトコール】 • 解析項目: 血漿中薬物濃度及び尿中排泄率 • 解析対象集団: 薬物動態解析には薬物動態解析対象集団を用いる. • 解析方法 – 求める薬物動態パラメータとその算出方法: AUCt 定量下限以上の値が得られた時間までの AUC(台形法による) – 統計解析方法: 血漿中薬物濃度及び薬物動態パラメータの要約統計量を投与量ごとに算出する. 【解析計画書】 • 血漿中濃度解析: 要約方法(平均値,標準偏差,幾何平均,変動係数,中央値,最大値,最小値,etc) • 薬物動態解析: – 求める薬物動態パラメータとその算出方法: AUCt 定量下限以上の値が得られた時間までの AUC(台形法による) – 統計解析方法: 血漿中薬物濃度及び薬物動態パラメータの要約統計量を投与量ごとに算出 する. 臨床試験において NCA に一般的に用いられているソフトウェアは Phoenix/WinNonlin(Phar-sight)や SAS(SAS Institute Inc.)である.
4.2 AUC 算出
NCA における AUC 算出の計算方法としては,一般的に各時点の濃度を直線で結んだ台形の面
積を用いる線形台形法(Linear trapezoidal method)(図 11)と各時点の濃度を指数関数で結んだ台
形の面積を用いる対数線形台形法(Log-linear trapezoidal method)(図 12)が用いられている.
線形台形法 AUCt= n X i=2 (Ci+ Ci−1) 2 (ti− ti−1) 対数線形台形法 AUCt= n X i=2 (Ci+ Ci−1) (ln Ci− ln Ci−1)(ti− ti−1) (Ci: 時点 i のときの濃度,ti: 時点 i のときの時間,n: 総時点数) 図 11. 線形台形法 図 12. 対数線形台形法
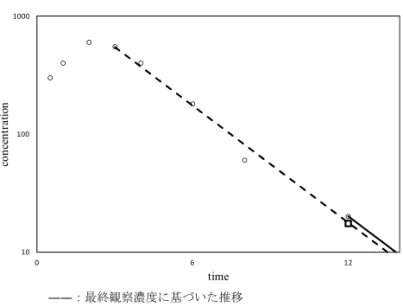
図 13. AUC∞算出時の外挿方法の比較
一般的に血漿中濃度は 1 次速度過程に従い消失すると仮定されるため,吸収相では線形台形法,
消失相では対数線形台形法による推定がよいと考えられている.そのため,tmaxまでの吸収相で
は線形台形法,tmax以降の消失相では対数線形台形法を用いる Linear Up Log Down という計算
方法が用いられる場合もある.また,tmaxが測定時点に依存するという問題等があることから, 全ての時点で線形台形法を用いる場合もある.第 I 相臨床試験の NCA 解析では線形台形法を用 いた場合と対数線形台形法を用いた場合で,開発の意思決定に影響するほどの違いが生じること は殆どなく,どの方法を用いるのか事前に規定しておけば問題ない. 無限時間まで外挿した AUC(AUC∞)の計算方法としては,最終観察濃度に基づく方法と,λz 推定に実行される線形回帰により推定した最終観察時点の濃度推定値に基づく方法の 2 通りが用 いられている(図 13).線形回帰により推定した濃度推定値が必ずしも信頼できるとは限らないた め,実際に得られた最終観察濃度を用いるほうが信頼できるという考え方と,最終観察濃度は測 定誤差も含んでいるため,最終観察時点の濃度推定値を用いた方が最終観察時点以降の AUC と してはより正確な値が求められるという考え方である.どちらの方法を用いるにしろ,どの方法 を用いるのか事前に規定しておけば問題ない. 最終観察濃度に基づいた AUC∞ AUC∞= AUCt+Ct ˆ λz λz推定に実行される線形回帰により推定した最終観察時点の濃度推定値に基づく AUC∞ AUC∞= AUCt+ ˆ Ct ˆ λz (Ct: 最終観察濃度が得られたときの濃度, ˆCt: 最終観察時点の推定濃度,ˆλz: 見かけの消失 速度定数推定値)
図 14. 最終観察時点までの AUC 最終時点の濃度が測定できない(定量下限未満: BLQ)場合の AUC の算出には,最終観察時点 までの濃度に基づき AUC を算出する方法と,最終時点の濃度を 0 として AUC を算出する方法が 用いられている.最終観察時点までの濃度に基づき AUC を算出する方法では最終時点の濃度が 定量下限を少し上回るか下回るかによって(図 14)最終時点のひとつ前の時点から最終時点までの 面積を含むか含まないかが変わるため,AUC が大きく異なってしまう.最終時点の濃度を 0 とし て AUC を算出する方法で最終時点の濃度が定量下限を少し下回る場合には,最終時点のひとつ 前の時点から最終時点の濃度 0 までの面積を AUC に含むため,最終観察時点までの濃度を用い た場合に比べて誤差は小さくなるが,最終時点よりも早くに濃度が 0 となる場合には,0 となっ た時点以降の面積も AUC に含むため,AUC を過大評価してしまうこととなる(図 15). 最終観察時点から求められる AUCtが AUC∞に比べて十分に大きくない場合には,消失相を 評価するのに十分な採血が行えておらず,求まった AUC∞は妥当な値とは考えられない.そのた め,「後発医薬品の生物学的同等性試験ガイドライン」(厚生労働省, 2012)では原則として AUCt が AUC∞の 80 %以上になる時点,消失半減期が非常に長い場合は,少なくとも 72 時間にわたっ て体液の採取を行う,と規定されている.そのため,解析計画書にも,1 − (AUCt/ AUC∞) > 0.2 となる場合,当該 AUC∞は集計に使用しない,等と規定し,妥当な AUC∞を用いて解析する必 要がある. 4.3 λz 算 出 λzを算出する時点群の選択は NCA において重要な検討事項の 1 つである.λzをどの時点群 を用いて算出するかによって λzの値が変わり(図 16),その結果 t1/2, AUC∞の算出にも影響が あるため,結果が恣意的ともなりうる.そのため,時点群の選択については事前に規定する必要 があり,WinNonlin では標準的に以下のルールを採用している.「BLQ を上回る濃度データのう ち,最後に測定された時点から連続した 3 点以上の Cmaxを含まない濃度データを用いる.tmax
図 15. 最終時点の濃度を 0 とした AUC 以降に BLQ を上回る濃度データが,3 点以上存在しない場合は,λz及び算出に λzを用いる薬物 動態学的パラメータの推定は実施しない.」.Cmaxを λzの算出に含まないのは,Cmaxが実測値 であり,採血時点によっては吸収相となる場合があるため,吸収相の Cmaxを含んで λzを算出す ると λzを真の値よりも小さく推定するためである. 図 16. λzを算出する時点群の選択 4.4 NCA のための採血時点
Cmax, tmaxを測定する測定時点の選択は NCA において重要な検討事項の 1 つである.Cmax,
か決まる.真の tmax付近で密に測定を行うことができれば真の Cmax, tmaxに近い値を得ること
ができるが,脈管外投与(血管,リンパ管以外への投与)で薬物動態の時間 − 濃度関係の予測を誤
り、真の tmax附近の測定を行うことできなかった場合には適切な Cmax, tmaxは得られず,tmax
のバラつきも大きくなりやすい. 4.5 生物学的同等性試験
NCA 解析を用いる試験の 1 つに生物学的同等性試験(以下,BE 試験)がある.BE 試験の目的 は 2 つの製剤(先発医薬品と後発医薬品,剤型変更等)の治療学的な同等性を保証することにあり, その方法はとしては主に 2 剤のバイオアベイラビリティの比較である.バイオアベイラビリティ とは有効成分の未変化体又は活性代謝物が体循環血中に入る速度と量であり,バイオアベイラビ リティが同等である製剤は生物学的に同等な製剤であると「後発医薬品の生物学的同等性試験ガ イドライン」(厚生労働省, 2012)に規定されている.また,BE 試験の同等性評価パラメータ及び その許容域は以下の様に規定されている.統計学的解析としては「原則として,tmaxを除くパラ メータでは対数正規分布することが多いので,対数変換をして解析し,90 %信頼区間(非対称,最 短区間)で生物学的同等性を評価する.」,「試験製剤と標準製剤の生物学的同等性評価パラメータ の対数値の平均値の差の 90 %信頼区間が,log(0.80) ∼ log(1.25) の範囲にあるとき,試験製剤と 標準製剤は生物学的に同等と判定する.」 【同等性評価パラメータ】 • 血液を採取体液とする単回投与試験
– AUCt及び Cmaxを生物学的同等性評価パラメータとする.Cmaxは実測値を用い,AUC
は台形法で計算した値を用いる. – AUC∞, tmax, MRT, kelなどは参考パラメータとする.ただし,作用発現時間の差が医薬 品の臨床的有用性に影響を与える可能性がある場合には,tmaxも同等性評価パラメータと する. 【生物学的同等の許容域】 • AUC 及び Cmaxが対数正規分布する場合 – 試験製剤と標準製剤のパラメータの母平均の比で表すとき 0.80 ∼ 1.25 • AUC 及び Cmaxが正規分布する場合 – 試験製剤と標準製剤のパラメータの母平均の差を標準製剤の母平均に対する比として表す とき −0.20 ∼ +0.20 5. 定常状態の解析 定常状態とは,投与間隔に比し半減期の長い薬物を反復投与した場合には体内の薬物は累積し て,単回投与時に比べてその血中濃度は上昇し,やがて,さらに投与を継続しても同じような血 中濃度推移を示すようになる.この時,血中濃度は定常状態に達したといわれる,と「医薬品の 臨床薬物動態試験について」(厚生労働省, 2001)では記載されている.反復投与を行った場合に どの程度定常状態に到達しているかは到達率の式を用いて求めることができる.
反復投与時の濃度 Cn(t)= C0·1 − e −n·kel·τ 1 − e−kel·τ · e −kel·t 定常状態の濃度 Css(t)= C0· 1 1 − e−kel·τ · e −kel·t 到達率 到達率 = 1 − e−n·kel·τ⇒ 1 − „ 1 2 « t t1/2 (n: 反復投与回数,kel: 消失速度定数,τ : 投与間隔,t1/2: 消失半減期,C0: 急速静注初回 投与直後の濃度,Cn(t): n 回反復投与時の時間 t のときの濃度,Css(t): 定常状態の時間 t のと きの濃度) 一般的には,半減期の 5 倍の時間が経過した場合を定常状態と見なし,反復投与試験で定常状 態の評価を行う時期等を検討している.実際の試験では定常状態の確認は薬物濃度推移の図を基 に目視で行っており,確認方法を SAP に記載しない場合もある. 半減期の 5 倍の時間が経過した時の到達率 到達率 = „ 1 −1 2 5« = 0.96875
定常状態及び単回投与時における AUC, Cmax, Cmin及び平均血中濃度(Cav)のそれぞれについ
て比(累積係数)を求め,それらが単回投与試験結果からの予測値と異なる場合には,次のことが 示唆される.実測累積係数が予測累積係数よりも大きい場合には,酵素やトランスポーターの阻 害やダウンレギュレーション,肝,腎などへの障害が示唆され,このような場合に蓄積性がある と言う.実測累積係数が予測累積係数よりも小さい場合には,酵素やトランスポーターの誘導な どが示唆される. 予測累積係数の算出について,Cmax及び Cminは単回投与時の濃度推移の重ね合わせ法による シミュレーションにより反復投与時の Cmax及び Cminを予測することで,求めることができる.
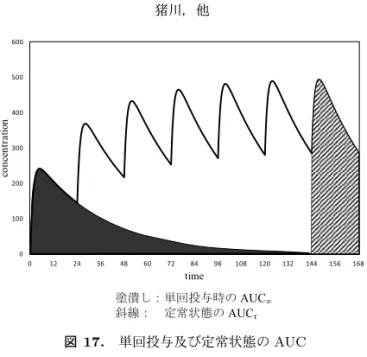
AUCτ及び Cavは薬物動態が線形であるならば単回投与時の AUC∞が反復投与時の AUCτと等
しいことから(図 17),以下の式により求まる.また,実測累積係数及び蓄積性評価も以下の式に
より算出できる. 【予測累積係数】
R(AUC)= AUC∞(day 1)/ AUCτ (day 1)
R(Cav)= Cav (day X) Cav (day 1) =AUC∞(day 1)/τ AUCτ (day 1)/τ 【実績累積係数】
R(AUC)= AUCτ (day X)/ AUCτ (day 1)
図 17. 単回投与及び定常状態の AUC
【蓄積性評価】
AI(AUC)= AUCτ (day X)/ AUC∞(day 1)
(day X : 定 常 状 態( 最 終 投 与 日 ),τ : 投 与 間 隔 ,AUC∞(day 1): 単 回 投 与 時 の AUC∞,
AUC∞(day X): 定常状態の AUC∞,AUCτ (day 1): 単回投与時の AUCτ,AUCτ (day X): 定常
状態の AUCτ,Cmax(day 1): 単回投与時の Cmax,Cmax(day X): 定常状態の Cmax,Cav (day 1):
単回投与時の Cav,Cav (day X): 定常状態の Cav) 6. 用量比例性 用量比例性とは,薬物動態が線形と見なせる場合に,薬物動態に関する速度(吸収速度,代謝速 度)が投与量に比例し,Cmax, AUC は投与量に比例することである.逆にいえば,線形性がある とは,薬物動態に関する速度(例えば,吸収速度,代謝速度など)が投与量に比例する場合をいう. また,広義には全ての薬物動態に関する速度過程が濃度に対して一次線形である場合,体内動態 に線形性があるという.このとき,血中濃度,AUC,Cmaxなどは投与量に比例する.即ち,横 軸に投与量,縦軸にこれらの薬物動態パラメータ値をとり,これらの関係をプロットしたとき, 回帰線は理論的に原点を通る直線となる.また,速度定数に関する薬物動態パラメータ(例えば, クリアランス,半減期,MRT など)は投与量によらず一定となる. Cmax∝ Dose AUC ∝ Dose 体内動態に線形性がある場合には,CL, Vd, Ka等は用量や投与回数(反復投与)によって変化し ないと見なすことができ,その場合を線形薬物動態といい,そうでない場合を非線形薬物動態と いう.薬物動態が線形と見なせない場合には,予測のためにより複雑な薬物動態モデルが必要と なる. 用量比例性があると見なせる場合には,投与量を 2 倍,3 倍にした場合に Cmax, AUC も 2 倍,
図 18. AUC と Cmax
3 倍となり,同じ投与量で 2 回,3 回に分けて投与した場合には 1 回で全量を投与した場合と比
較して AUC は同じとなるが,Cmaxは異なる(図 18).
用量比例性の評価方法としては,直線回帰分析,分散分析,パワーモデルによる解析が用いら れている.
直線回帰分析を用いた評価方法では AUCi= α + β · Dosei+εi(AUCi: 被験者 i の AUC,
Dosei: 被験者 i の投与量,εi: 被験者 i の個体間変動)において α = 0 の場合に AUC = β · Dose
と用量比例的となることから,回帰分析の検定で有意差を認めない場合に,帰無仮説 H0: α = 0 が棄却できない,すなわち用量比例的ではないとは言えないことを根拠に線形とみなしている. 【直線回帰分析】 AUCi= α + β · Dosei+εi H0: α = 0 (AUCi: 被験者 i の AUC,Dosei: 被験者 i の投与量,εi: 被験者 i の個体間変動)
分散分析を用いた評価方法では AUC が用量比例的であれば AUC と Dose の比が一定となるこ とから,各投与量群の用量補正した AUC の分散分析の検定結果に有意差を認めない場合に,下 記の帰無仮説が棄却できない,すなわち用量比例的ではないとは言えないことを根拠に線形とみ なしている. 【分散分析】 H0: AUCdose 1 Dose 1 = AUCdose 2 Dose 2 = AUCdose 3 Dose 3 · · · (AUCdose X: Dose X を投与した群の AUC 平均)
と用量比例的となることから,検定で有意差を認めない場合には帰無仮説 H0: β = 1 が棄却でき
ない,すなわち用量比例的ではないとは言えないことを根拠に線形とみなしている.検定の際は
対数変換を行う(log(AUCi) = log(α) + β · log(Dosei) + εi)ことで容易に実行することができる。
【パワーモデル】
AUC = α · Doseβ
log(AUCi) = log(α) + β · log(Dosei) + εi
H0: β = 1 (AUCi: 被験者 i の AUC,Dosei: 被験者 i の投与量,εi: 被験者 i の個体間変動) 参考文献 加藤隆一 (2010). 臨床薬物動態学: 臨床薬理学・薬物療法の基礎として(第 4 版). 南江堂(東京). 厚生労働省 (2001). 医薬品の臨床薬物動態試験について 厚生労働省 (2012). 後発医薬品の生物学的同等性試験ガイドライン 九川文彦 (2013). 徹底解説 薬物動態の数学: 微積分と対数,非線形(第 2 版). 廣川書店(東京). Malcolm Rowland and Thomas N. Tozer. (2012). Clinical pharmacokinetics and
pharmacody-namics: concepts and applications (4th edition). Lippincott Williams & Wilkins (Pennsyl-vania, USA).
増原慶壮, 松本宜明, 木島慎一, 高橋晴美, 緒方宏泰 (2007). 臨床薬物動態学: 薬物治療の適正化の ために(第 2 版). 丸善出版(東京).
付表: http://pub.maruzen.co.jp/book magazine/rinsho yakubutsu/fuhyo/
Michael E. Winter (2009). Basic clinical pharmacokinetics (5th edition). Lippincott Williams & Wilkins (Pennsylvania, USA).
臨床薬物動態試験・薬物相互作用ガイドライン検討班 (2003). 医薬品の臨床薬物動態試験: 通知 解説. じほう(東京).
杉山雄一, 楠原洋之 (2008). 分子薬物動態学. 南江堂(東京). 高田寛治 (2002). 薬物動態学: 基礎と応用(第 2 版). じほう(東京).