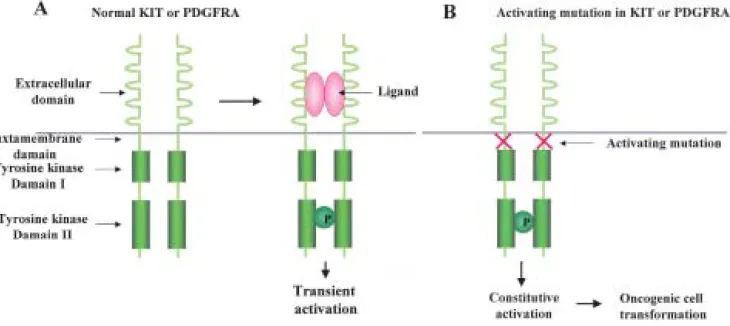
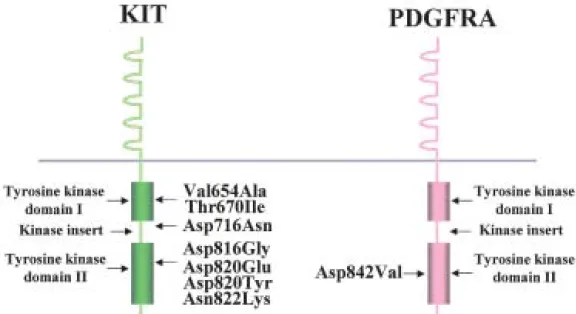
stromal tumor was introduced
11
0
0
全文
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
図
関連したドキュメント
The mGoI framework provides token machine semantics of effectful computations, namely computations with algebraic effects, in which effectful λ-terms are translated to transducers..
We have found that the model can account for (1) antigen recognition, (2) an innate immune response (neutrophils and macrophages), (3) an adaptive immune response (T cells), 4)
All (4 × 4) rank one solutions of the Yang equation with rational vacuum curve with ordinary double point are gauge equivalent to the Cherednik solution.. The Cherednik and the
A lemma of considerable generality is proved from which one can obtain inequali- ties of Popoviciu’s type involving norms in a Banach space and Gram determinants.. Key words
discrete ill-posed problems, Krylov projection methods, Tikhonov regularization, Lanczos bidiago- nalization, nonsymmetric Lanczos process, Arnoldi algorithm, discrepancy
[Mag3] , Painlev´ e-type differential equations for the recurrence coefficients of semi- classical orthogonal polynomials, J. Zaslavsky , Asymptotic expansions of ratios of
As already mentioned, the above selection has to be regarded as a way to reduce complexity, however, pursuing the objective of designing models suitable to provide a
Jin [21] proved by nonstandard methods the following beautiful property: If A and B are sets of natural numbers with positive upper Banach density, then the corresponding sumset A +
