糖尿病治療薬としての11β-hydroxysteroid
dehydrogengase type 1 阻害剤の合成と構造活性相
関に関する研究
著者
小池 貴徳
学位授与機関
Tohoku University
学位授与番号
11301甲第19325号
URL
http://hdl.handle.net/10097/00127870
糖尿病治療薬としての
11β-hydroxysteroid dehydrogenase type 1 阻害剤の合成と
構造活性相関に関する研究
東北大学大学院農学研究科
生物産業創成科学専攻
小池 貴徳
指導教員
桑原 重文 教授
1 目次 序論 6 本論 第一章 新規トリアゾール誘導体の合成 第一節 リード化合物とその課題 11 第二節 合成方針 12 第三節 化合物の合成 13 第四節 第一章のまとめ 20 第二章 構造活性相関 第一節 チオフェン部の構造活性相関 21 第二節 トリアゾール環3 位の構造活性相関 23 第三節 チオフェン環上置換基の構造活性相関 25 第四節 トリアゾール環4 位の構造活性相関 28 第五節 第二章のまとめ 30 第三章 化合物 3 の抗糖尿病作用の評価、及びヒト 11β-HSD1 阻害活性、種差に関する考 察 第一節 化合物3 の in vivo 評価 32 第二節 化合物3 のヒト 11β-HSD1 阻害活性及び種差に関する考察 35 第三節 第三章のまとめ 37
2 第四章 化合物3 の合成法の改良 第一節 化合物3 の合成① 39 第二節 化合物3 の合成② 41 第三節 第四章のまとめ 45 総括 46 実験の部 51 参考文献 89 論文目録 92 謝辞 93
3 略語表
本論文において以下に示す略語及び略号を用いた。 AcOH acetic acid
bid bis in die
BMI body mass index
Bn benzyl
Boc tertiary-butoxycarbonyl
CAS No. Chemical Abstracts Service Registry Number CLint hepatic intrinsic clearance
c-Bu cyclobutyl CDI 1,1-carbonyldiimidazole c-Hex cyclohexyl CN cyano c-Pen cyclopentyl c-Pr cyclopropyl DIPEA N,N-diisopropylethylamine DMF N,N-dimethylformamide DMSO dimethylsulfoxide Et ethyl Et3N triethylamine
4 Et2O diethyl ether
EtOAc ethyl acetate
EtOH ethanol
HOBt 1-hydroxybenzotriazole
HOMA-IR homeostasis model assessment of insulin resistance HTRF homogeneous time-resolved fluorescence method HTS high-throughput screening
HRMS high-resolution mass spectrometry IC50 50% inhibitory concentration
iPr isopropyl
LC-MS liquid chromatography–mass spectrometry
Me methyl
MeI iodomethane
MeOH methanol
MeOTf methyl trifluoromethanesulfonate
NADP nicotinamide-adenine dinucleotide phosphate
NBS N-bromosuccinimide
NCS N-chlorosuccinimide
NIS N-iodosuccinimide
5 NMR nuclear magnetic resonance
n-Pr normal propyl
po per os
r.t. room temperature
t-Bu tertiary butyl
THF tetrahydrofuran
WSCD water-soluble carbodiimide
11-HSD1 11-hydroxysteroid dehydrogenase type 1 11β-HSD2 11β-hydroxysteroid dehydrogenase type 2
6 序論 本研究の背景 糖尿病とは 糖尿病とは、膵臓のβ細胞からのインスリン分泌不全、または抹消組織におけるインス リン抵抗性に起因する、インスリン作用不足の結果として高血糖が慢性的に持続する代謝 疾患である1)。日本では糖尿病が強く疑われる人が約1000 万人、糖尿病の可能性が否定で きない人が約1000 万人で、合計すると 2000 万人と報告されており(平成 28 年厚生労働省 調査)、「国民病」の1つと言えよう。糖尿病は発生原因により大きく 2 つに分類されてい る 2)。膵臓のβ細胞の破壊や自己免疫疾患によるインスリン欠乏によって発症する 1 型糖 尿病と、遺伝的要因や、肥満、過食、運動不足、高脂肪食などの環境要因によるインスリ ン抵抗性、インスリン分泌低下により発症する 2 型糖尿病であり、日本では 2 型糖尿病患 者が約 95%である。糖尿病にはほとんどの場合自覚症状が無く、何らかの自覚症状が出る 頃には糖尿病は進行しており、治療が遅れると糖尿病合併症を発症する。特に神経障害、 網膜症、腎症が糖尿病の3 大合併症と呼ばれており、早期の診断、治療が必要である。 糖尿病治療薬 糖尿病の治療は食事療法と運動療法が基本であるが、食事療法、運動療法では血糖コン トロールが不十分である場合には薬物療法を行う。数多くの 2 型糖尿病治療薬が研究され ており 3)、インスリン分泌促進剤やインスリン抵抗性改善薬が開発され種々の糖尿病治療
7 薬が上市されている。Table 1 に経口糖尿病治療薬の一覧を示す。しかしながら、限られた 薬理学的効果や有害な副作用のために、多くの 2 型糖尿病患者では十分な血糖コントロー ルは達成されていない。そのため、新たな作用機序を有する薬剤の開発が求められている。 Table 1. 経口糖尿病治療薬 分類 作用機序 副作用例 一般名 イ ン ス リ ン 分 泌 促 進 型 SU 剤 膵 β 細胞に作用し、 インスリンを分泌 低血糖 グリベンクラミ ド等 DPP-IV 阻害薬 インクレチンの分解 を抑制し、インスリ ン分泌を増強 他 剤 併 用 時 の 低 血 糖 シタグリプチン 塩酸塩等 非 イ ン ス リ ン 分 泌 促 進 型 α-グルコシダーゼ 阻害薬 腸管内で糖質のブド ウ糖への分解阻害 放屁、お腹の張り ボグリボース等 ビグアナイド薬 糖新生抑制 乳酸アシドーシス メトホルミン塩 酸塩 ブホルミン塩酸 塩 チアゾリジン薬 PPARγ 活性化による インスリン抵抗性改 善 浮腫、心不全、体重 増加 ピオグリタゾン 塩酸塩 SGLT2 阻害薬 血液中の糖分を尿中 に排泄することによ り血糖を低下 頻尿、多尿による脱 水症状、尿路・性器 の感染症 イプラグリフロ ジ ン L- プ ロ リ ン等 11-HSD1 11β-Hydroxysteroid dehydrogenase はグルココルチコイドの変換を触媒する酵素であり、2
8
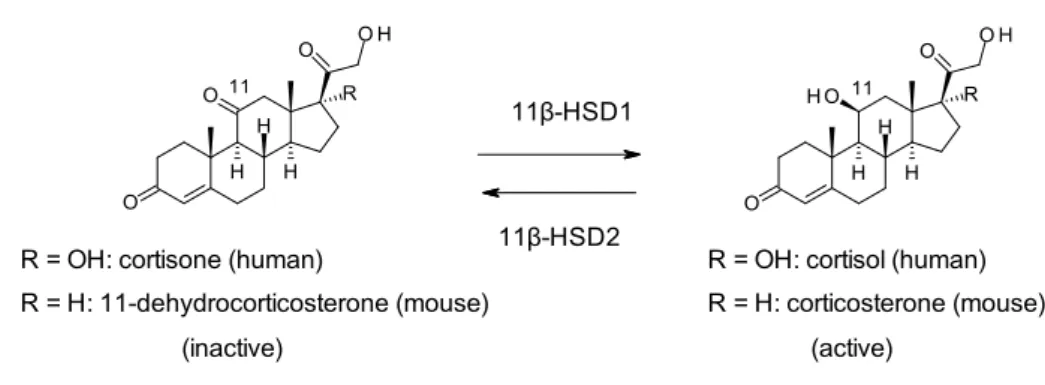
種類のアイソザイムが同定されている(Figure 1)。11-hydroxysteroid dehydrogenase type 1 (11-HSD1)は肝臓や脂肪組織において不活性型グルココルチコイド(ヒト: cortizone, マ ウ ス: 11-dehydrocorticosterone)を活性型グルココルチコイド (ヒト: cortisol, マウス: corticosterone)に変換する酵素である。一方で、11β-hydroxysteroid dehydrogenase type 2(11β-HSD2)は腎臓において活性型グルココルチコイドを不活性型グルココルチコイドに変換す る。
11
R = OH: cortisol (human) R = H: corticosterone (mouse)
11
(inactive) (active)
R = OH: cortisone (human)
R = H: 11-dehydrocorticosterone (mouse)
11β-HSD1
11β-HSD2
Figure 1. Interconversion of glucocorticoids in humans and mice.
脂肪組織におけるグルココルチコイドの異常な活性化と糖尿病との関係が明らかになっ ている 4)。また、肥満患者の脂肪組織における11β-HSD1 活性の亢進5)、11β-HSD1 活性と BMI、HOMA-IR、空腹時血糖との相関が報告されている6)。加えて、脂肪組織選択的に 11β-HSD1 を過剰発現させたトランスジェニックマウスでは、脂肪組織における活性型グルコ コルチコイドレベルが上昇し、インスリン抵抗性および高脂血症を示すことが知られてい る 7,8)。従って、11β-HSD1 阻害剤は、不活性型グルココルチコイドから活性型グルココル チコイドへの変換を抑制することにより、活性型グルココルチコイドによる高血糖、イン スリン抵抗性および高脂血症などの代謝異常を改善すると期待される(Figure 2)。また、
9 腎臓における 11β-HSD2 の阻害が高血圧を惹起することが知られていることから 9)、糖尿 病治療を目的とした11β-HSD1 阻害剤は 11β-HSD2 に対して十分な乖離を示す必要がある。 現在までに種々の11β-HSD1 阻害剤が報告されており10)、いくつかの化合物11–16)につい て臨床試験が実施されているが(Figure 3)、いずれの化合物も未だ上市には至っておらず 新たなタイプの HSD1 阻害剤が必要であると考えられる。従って、著者は新規な 11β-HSD1 阻害剤の創製を目指し本研究に着手した。 Figure 2. 作用機序仮説 MK-0916 AMG-221 BI-135585 BVT-3498 HSD-016 PF-915275
10 本研究の目的 本研究では、2 型糖尿病の治療薬としての有用性が期待される新規 11β-HSD1 阻害剤の 創製を目的に研究を行った。目標とする化合物のプロファイルとして以下の項目を設定し た。 ① ヒト 11β-HSD1 に対して強力な阻害活性を示す ② マウス糖尿病モデルにおいて、経口投与で抗糖尿病作用を示す 研究方針として、HTS によりヒット化合物を見出し、ヒット化合物からの合成展開によ り、ヒト11β-HSD1、ヒト 11β-HSD2、マウス肝ミクロソーム代謝安定性に関する構造活性 相関を明らかにし、糖尿病モデルマウスにおいて有効性を示す化合物の創出を目指すこと とした。
11 本論 第一章 新規トリアゾール誘導体の合成 第一節 リード化合物とその課題 2 型糖尿病の新しいメカニズムの薬剤として、新規の 11β-HSD1 阻害剤を得るためにア ステラス製薬所有化合物ライブラリーを用いたHTS を行い、新規 11β-HSD1 阻害剤として 化合物1 を見出した。初期の研究において、特許性を有する化合物を得ることを目的とし、 1 のリンカー部、クロロベンゼン部、トリアゾール環の 4 位と 5 位の置換基の変換行うこ
とでリード化合物2 を得た(unpublished data; Figure 4)。2 はヒト 11β-HSD2 に対する阻害
活性は有さないものの、ヒト11β-HSD1 阻害活性は中程度にとどまり(IC50 = 23 nM)、マウ ス代謝安定性も不十分であった(CLint= 1122 mL/min/kg)。従って、臨床試験へと進める前 提として、ヒト 11β-HSD1 阻害活性の増強とともに、糖尿病モデルマウスでの有効性を担 保するためのマウス肝ミクロソーム安定性改善が必要である。これら 2 点の課題解決を目 指して、2 の構造変換を行った。 4 5 human 11β-HSD1: IC50 = 23 nM human 11β-HSD2: IC50 = >10000 nM mouse CLint = 1122 mL/min/kg
1 2
12 第二節 合成方針 著者は以下2点の課題解決のための合成方針を策定した(Figure 5)。 ヒト11β-HSD1阻害活性の向上 マウス肝ミクロソーム安定性の改善 ヒト11β-HSD1阻害活性の向上に関しては、11β-HSD1の生体内リガンドがステロイドで あることから、11β-HSD1タンパクは脂溶性の高いものや嵩高いものを好むと仮定し、ト リアゾール環3、4位の脂溶性の増大や、嵩高い置換基の導入を中心に構造変換を行う合成 方針を立てた。 一方で、マウス代謝安定性の改善には化合物全体の脂溶性の低下か代謝部位のブロック が必要であると考えられる。前者に関してはチオフェン環を含窒素ヘテロ環に変換するこ とで脂溶性を下げる方針とした。後者についてはチオフェン環が2の主代謝部位であると 仮定し、チオフェン環の酸化代謝をブロックするために電子吸引性置換基を中心に置換基 導入を行う合成方針を策定した。 3 4 ・脂溶性の増大 ・嵩高い置換基の導入 ・含窒素ヘテロ環の導入による脂 溶性の低下 ・置換基導入によるチオフェンの酸化 代謝のブロック 青:ヒト11β-HSD1阻害活性向上を指向 赤:マウス肝ミクロソーム安定性改善を指向 human 11β-HSD1: IC50 = 23 nM mouse CLint = 1122 mL/min/kg
2
13 第三節 化合物の合成 化合物 2 のチオフェン部及びトリアゾール環上置換基の変換によるヒト 11β-HSD1 阻害 活性の向上、マウス代謝安定性の改善を目的として化合物3–32 を合成した。評価化合物の 一覧をTable 2 に示す。 まず、チオフェン環の酸化代謝の抑制によるマウス代謝安定性の改善を目的として、チ オフェン環にフルオロ基及びトリフルオロメチル基が導入された化合物3、4、5 を合成し た。まず、3 の合成を Scheme 1 に示す。カルボン酸 33 のメチル化を行いエステル 34 を 得た後、34 をエタノール中でヒドラジン一水和物と加熱することによりヒドラジド 35 と した。次に、35 と 36 を MeOTf を用いて環化させることでトリアゾール 3’を得た。最後 に、3’を 4 M HCl/EtOAc を用いて造塩反応を行い、3 を合成した。 次に、化合物4 の合成を Scheme 2 に示す。4 は 3 と同様の方法で合成した。エステル 37 をエタノール中でヒドラジン一水和物と加熱することによりヒドラジド 38 とした後、38 と36 を MeOTf を用いて環化させることにより 4 を得た。 続いて、化合物5 の合成を Scheme 3 に示す。まず、ニトリル 39 のα位をジメチル化し て40 を得た後、40 のシアノ基を加水分解することによりカルボン酸 41 を得た。41 とヒド ラジンをWSCD、HOBt を用いて縮合し、生成したヒドラジド 42 とアミド 36 を MeOTf を 用いて環化することによりトリアゾール5 を得た。
14
Table 2. Structures of compounds 2–32
2–32 Compound R1 R2 R3 2 2-thienyl c -Pr, c -Pr 3a 2-(3-F-5-CF 3-thienyl) c -Pr c -Pr 4 2-(5-CF3-thienyl) c -Pr c -Pr 5 2-(3-F-thienyl) c -Pr c -Pr 6a 2-thienyl c -Pr n-Pr 7 2-thienyl c -Pr cyclopropylmethyl 8 2-thienyl c -Pr t-Bu 9 2-thienyl c -Pr c-Bu 10 2-thienyl c -Pr c-Pen 11 2-thienyl c -Pr c-Hex 12a 2-(5-Br-thienyl) Me c -Pr 13a 2-(5-Br-thienyl) Et c -Pr 14a 2-(5-F-thienyl) c -Pr c -Pr 15 2-(4-Br-thienyl) c -Pr c -Pr 16 2-(3-Cl-thienyl) c -Pr c -Pr 17 2-pyrazyl c -Pr c -Pr 18 2-(3-F-5-Cl-thienyl) c -Pr c -Pr 19 2-(5-Cl-thienyl) c -Pr c -Pr 20 2-(3,5-di-Cl-thienyl) c -Pr c -Pr 21 2-(5-Br-thienyl) c -Pr c -Pr 22 2-(5-Me-thienyl) c -Pr c -Pr 23 2-(5-CN-thienyl) c -Pr c -Pr 24a 1-pyrrolyl c -Pr c -Pr 25 2-thienyl c -Pr H 26a 2-(5-Br-thienyl) n-Pr c -Pr 27a 2-(5-Br-thienyl) i-Pr c -Pr 28 2-(5-Br-thienyl) cyclopropylmethyl c -Pr 29a 2-(5-Br-thienyl) Bn c -Pr 30 2-(5-Br-thienyl) phenethyl c -Pr 31a 2-pyridyl c -Pr c -Pr 32a 4-pyridyl c -Pr c -Pr a) Hydrochloride salt.
15 3' 34 35 36 HCl b c d a 3 33
Scheme 1. Synthesis of 3. Reagents and conditions: (a) MeI, K2CO3, DMF, r.t., 15 h; (b) hydrazine monohydrate, EtOH, 80°C, 24 h; (c) 36, MeOTf, 60°C, 30 min then Et3N, toluene, 60°C, 24 h, 90°C, 9 h, 110°C, 15 h; (d) 4 M HCl/EtOAc, EtOAc.
4
37 38
a b
Scheme 2. Synthesis of 4. Reagents and conditions: (a) hydrazine monohydrate, EtOH, 80°C, 24 h;
(b) 36, MeOTf, 60°C, 30 min then Et3N, toluene, 70°C, 17 h, 110°C, 7 h.
a d 42 c 5 b 39 40 41
Scheme 3. Synthesis of 5. Reagents and conditions: (a) MeI, NaH, DMF, r.t., 20 min; (b) KOH,
ethylene glycol, 190°C, 2.5 h; (c) hydrazine monohydrate, WSCD·HCl, HOBt·H2O, CH2Cl2, r.t.,
overnight; (d) 36, MeOTf, Et3N, toluene, 60°C, 2 d, 100°C, 2 h.
また、化合物2、及びヒト 11β-HSD1 阻害活性の向上、マウス代謝安定性の改善を目的と
16
をScheme 4 に示す。3 と同様の方法でヒドラジド A とアミド B を MeOTf を用いて環化す
ることで2、6–17 を合成した。
A B 2, 6–17
a
Scheme 4. Synthesis of 2, 6–17. (a) 36, MeOTf, Et3N, toluene, 60 to 110°C.
次に、チオフェン環の酸化代謝の抑制によるマウス代謝安定性の改善を目的として、チ オフェン環に各種置換基を導入した化合物18–23を合成した(Scheme 5)。まず、NCSを 用いて化合物5、2、16の塩素化を行い、18、19、20を得た。また、2をNBSを用いて臭素 化することで21を合成し、続いて、21をn-BuLiとDMFで処理することによりアルデヒド43 を得た。さらに、43をWolff-Kishner還元により22へと変換した。また、2をNISによりヨウ 素化し、続いてCuCNと反応させることでニトリル23を合成した。 続いて、脂溶性の低下によるマウス代謝安定性の改善を目的として、チオフェン環をピ ロール環に変換した化合物24 を合成した(Scheme 6)。まず、化合物 45 と 36 を MeOTf を 用いて環化することによりトリアゾール46 を合成し、続いて、46 を 4 M HCl/EtOAc で処 理 す る こ と に よ り Boc 基 を 脱 保 護 し ア ミ ン 47 を 得 た 。 最 後 に 、 47 を 2,5-dimethoxytetrahydrofuran と反応させることでピロール 24 を合成した。 また、トリアゾール環5 位の置換基の必要性を明らかにするために化合物 25 を合成し
た(Scheme 7)。化合物 48 を塩化チオニルと反応させ、続いて formyl hydrazine を用いて
17 a c b e d f 21 43 22 44 23 18 R1= F, R2 = Cl 19 R1= H, R2 = Cl 20 R1= R2 = Cl 5 R1= F, R2 = H 2 R1= R2 = H 16 R1= Cl, R2 = H
Scheme 5. Synthesis of 18–23. Reagents and conditions: (a) NCS, AcOH, 50–80°C, 2 h–3 d; (b)
NBS, AcOH, 80°C, 2 h; (c) n-BuLi, N,N,N',N'-tetramethyl ethylenediamine, DMF, THF, –78°C, 45 min; (d) hydrazine monohydrate, KOH, diethylene glycol, 170°C, 4 h; (e) NIS, AcOH, r.t.,
overnight; (f) CuCN, pyridine, 115°C, 11.5 h.
Boc Boc
b c
a
45 46 47 24
Scheme 6. Synthesis of 24. Reagents and conditions: (a) 36, MeOTf, Et3N, toluene, 60 to 100°C, 3 h; (b) 4 M HCl/EtOAc, EtOH, 50°C, 8 h; (c) 2,5-dimethoxytetrahydrofuran, AcOH, CHCl3, 70°C, 24 h.
5
48 25
a, b
Scheme 7. Synthesis of 25. Reagents and conditions: (a) SOCl2, DMF, CHCl3, 60°C, 1 h; (b)
18 さらに、脂溶性及び嵩高さの増大によるヒト 11β-HSD1 阻害活性の向上を目的として、 トリアゾール環の4 位に各種の炭化水素基が導入された化合物 26–30 を合成した(Scheme 8)。まず、49 と cyclopropanecarbonyl chloride を縮合させることで化合物 50 を得た。続い て、50 を trifluoromethanesulfonic anhydride を用いて環化反応を行い、オキサジアゾール 51 を合成した。次に、51 を臭素化することで化合物 52 を得た。最後に、マイクロウェーブ反 応装置を用いて 52 と種々のアミンを反応させることによりトリアゾール 26–30 を合成し た。 4 b d c 49 50 51 52 26 R = n-Pr 27 R = i-Pr 28 R = cyclopropylmethyl 29 R = Bn 30 R = phenethyl a
Scheme 8. Synthesis of 26–30. Reagents and conditions: (a) cyclopropanecarbonyl chloride, Et3N, CH2Cl2, r.t., overnight; (b) trifluoromethanesulfonic anhydride, pyridine, CH2Cl2, r.t., 3 d; (c) NBS, AcOH, r.t.; (d) amine, AcOH, 170°C, 40–60 min (microwave).
最後に、脂溶性の低下によるマウス代謝安定性の改善を目的として、チオフェン環をピ
リジン環に変換した化合物 31、32 を合成した(Scheme 9)。まず、化合物 53、54 を
cyclopropanecarbonyl chloride と縮合させることにより化合物 55、56 を得た。続いて、55、
19 ブ反応装置を用いて 57、58 とシクロプロピルアミンを反応させることにより 31、32 を合 成した。 b c 53 R = 2-pyridyl 54 R = 4-pyridyl 57 R = 2-pyridyl 58 R = 4-pyridyl 31 R = 2-pyridyl 32 R = 4-pyridyl 55 R = 2-pyridyl 56 R = 4-pyridyl a
Scheme 9. Synthesis of 31 and 32. Reagents and conditions: (a) cyclopropanecarbonyl chloride,
Et3N, CH2Cl2, 0°C to r.t., 3–14 h; (b) phosphorus oxychloride, reflux, 2–8 h; (c) cyclopropylamine, AcOH, 175°C, 40 min (microwave).
20 第四節 第一章のまとめ 著者は、2 型糖尿病の新しいメカニズムの薬剤として、新規な 11β-HSD1 阻害剤の創製を 目指して研究に着手した。まず、アステラス製薬所有化合物ライブラリーを用いたHTS を 行い、新規 11β-HSD1 阻害剤として化合物 1 を見出した。次に、特許性の取得を目的とし た 1 からの合成展開によりリード化合物 2 を見出したが、2 はヒト 11β-HSD2 に対する阻 害活性は有さないものの、ヒト11β-HSD1 阻害活性は中程度にとどまり(IC50 = 23 nM)、マ ウス代謝安定性も不十分であった(CLint= 1122 mL/min/kg)。従って、ヒト 11β-HSD1 阻害 活性の向上、マウス肝ミクロソーム安定性の改善の 2 点の課題解決を目指し 2 の各種誘導 体の合成を行った。ヒト11β-HSD1 阻害活性の向上に関しては、トリアゾール環 3、4 位の 脂溶性の増大や嵩高い置換基の導入を中心に構造変換を行った。また、マウス代謝安定性 の改善については、チオフェン環を含窒素ヘテロ環に変換することによる脂溶性の低減、 及びチオフェン環の酸化代謝を抑制するために電子吸引性置換基を中心とした、チオフェ ン環への置換基導入を行った。評価化合物2–32 は Scheme 1–9 に記載した方法により調製 した。
21 第二章 構造活性相関
第一節 チオフェン部の構造活性相関
合成した化合物のin vitro ヒト 11β-HSD1、11β-HSD2、マウス 11β-HSD1 阻害活性につい て、大腸菌に発現させたヒト組み換え11β-HSD1 タンパク(residues 24–292 with His-tag at N-terminal)と、HEK-293*に発現させたヒト組み換え全長 11β-HSD2 タンパクをそれぞれ精製、
使用し、HTRF®を用いて測定した。マウス代謝安定性はマウス肝ミクロソームを用いて測
定した。また、CLogP 値は ACD LogP 予測ソフト(ACD/Percepta)17)を用いて計算した。
最初に、脂溶性の低減を指向し、チオフェン環を含窒素ヘテロ芳香環に変換した類縁体 の評価を行った(Table 3)。参照データとして化合物 2 のデータを示した。2 と比較して、ピ リジン体31、32 はヒト 11β-HSD1 阻害活性は減弱し、ピラジン体 17 はヒト 11β-HSD1 阻害 活性が大幅に減弱した。また、ピロール体24 は 2 と比較してヒト 11β-HSD1 阻害活性は約 6 分の 1 に減弱し、末端チオフェン部位では含窒素ヘテロ芳香環の許容性が低いことを示 唆した。一方で、2 と比較して 24 はマウス肝ミクロソーム安定性が改善した。両化合物の CLogP 値の比較の結果、脂溶性の低下により 24 のマウス肝ミクロソーム安定性が改善さ れたと推察される。以上の結果より、R 部はチオフェン環が最も適切な置換基であると結 論付けた。 ________________________________________ *HEK-293:ヒト胎児の腎由来の細胞株。培養がしやすく遺伝子導入が容易なため、組み換えタンパク質生成 に用いられる。
22
Table 3. Conversion of the thienyl moiety
17, 24, 31, 32 Compound R Human 11β-HSD1 IC50 (nM) Human 11β-HSD2 IC50 (nM) Mouse CLint (mL/min/kg) CLogP 2 23 >10000 1122 2.95 31a 591 NTb NTb 2.08 32a 343 NTb NTb 2.08 17 >3000 NTb NTb 1.52 24a 134 >30000 368 2.39 a) Hydrochloride salt. b) Not tested.
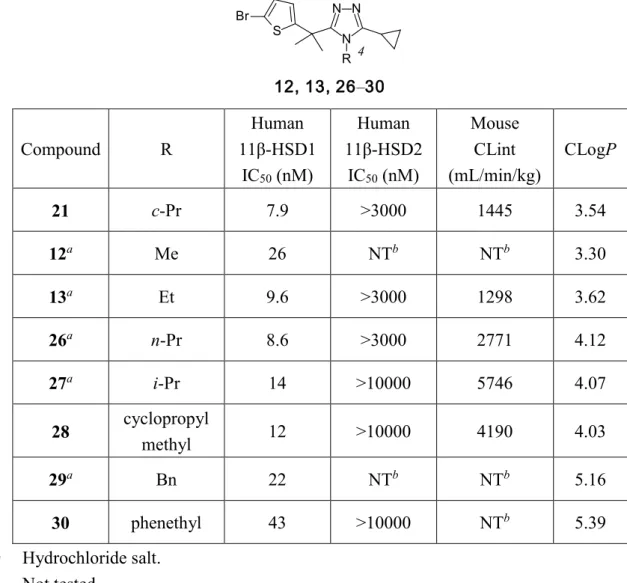
23 第二節 トリアゾール環3 位の構造活性相関 次に、脂溶性及び嵩高さの増大によるヒト 11β-HSD1 阻害活性の向上を目的とし、トリ アゾール環3 位の置換基変換を行った場合の評価結果を Table 4 に示す。置換基を除去した 化合物25 はヒト 11β-HSD1 阻害活性が消失し、トリアゾール環 3 位には置換基が必要であ ることを示唆した。シクロプロピル基を n-プロピル基に変換した化合物 6、シクロプロピ ルメチル基に変換した化合物7 は 2 と比較して、ヒト 11β-HSD1 阻害活性が約 2 分の 1 に 減弱した。一方で、tert-ブチル体 8、シクロブチル体 9、シクロペンチル体 10、シクロへキ シル体11 は 2 と比較してヒト 11β-HSD1 阻害活性は若干向上し、トリアゾール環 3 位は嵩 高い置換基が好ましいことが示唆された。また、化合物6–11 は 2 と比較してマウス肝ミク ロソーム安定性が低下した。CLogP 値の比較により、脂溶性の増大の結果、6–11 はマウス 代謝安定性が低下したと考えられる。なお、ヒト 11β-HSD2 阻害活性に関しては、いずれ の化合物も阻害活性を示さなかった。以上の結果より、トリアゾール環 3 位はシクロプロ ピル基が最も適切な置換基であることが示された。
24
Table 4. Conversion of the 3-position substituent of 1,2,4-triazole
6–11, 25 3 Compound R Human 11β-HSD1 IC50 (nM) Human 11β-HSD2 IC50 (nM) Mouse CLint (mL/min/kg) CLogP 2 c-Pr 23 >10000 1122 2.95 25 H >3000 NTb NTb 2.46 6a n-Pr 51 >10000 1705 3.53 7 cyclopropyl methyl 51 >10000 2313 3.38 8 t-Bu 16 >3000 2578 4.05 9 c-Bu 13 >3000 1460 3.42 10 c-Pen 14 >3000 3024 3.99 11 c-Hex 10 >3000 5862 4.37 a) Hydrochloride salt. b) Not tested.
25 第三節 チオフェン環上置換基の構造活性相関 続いて、チオフェン環の酸化代謝を抑制するために、化合物2 のチオフェン環に電子吸 引性基等の置換基を導入した場合の評価結果をTable 5 に示す。チオフェン環 5 位をハロ ゲン基に置換したフルオロ体14、クロロ体 19、ブロモ体 21 は 2 と比較してヒト 11β-HSD1 阻害活性は向上したが、マウス肝ミクロソーム安定性を改善する効果は得られな かった。また、5-メチル体 22 は 2 と比較して同等のヒト 11β-HSD1 阻害活性を示した が、マウス肝ミクロソーム安定性は約2 分の 1 に低下した。一方で、5-シアノ体 23 は 2 と比較してヒト11β-HSD1 阻害活性は約 4 分の 1 に減弱したものの、マウス肝ミクロソー ム安定性は約3 倍向上した。2 と 23 の CLogP 値の比較により、この代謝安定性の改善は 脂溶性の低下によるものと推察している。興味深いことに、5-トリフルオロメチル体 4 は 2 と同等のヒト 11β-HSD1 阻害活性を示し、2 と比較して CLogP 値が増大しているにも関 わらずマウス肝ミクロソーム安定性は約3 倍向上した。この結果から、トリフルオロメチ ル基の導入によるマウス代謝安定性の改善は、チオフェン環の酸化代謝の抑制によるもの であると推察された。4-ブロモ体 15、3-フルオロ体 5、3-クロロ体 16 は 2 と比較してヒト 11β-HSD1 阻害活性は向上したが、マウス肝ミクロソーム安定性の改善は示さなかった。 特に3-クロロ体 16 はマウス代謝安定性の大幅な低下を示し、クロロ基の導入がチオフェ ン環3 位において好ましくないことが示唆された。3,5-二置換誘導体に関しては、3-フル オロ-5-クロロ体 18、3,5-ジクロロ体 20 は 2 と比較してヒト 11β-HSD1 阻害活性が向上し た。しかしながら、両化合物ともにマウス肝ミクロソーム安定性は低下し、特に3,5-ジク
26
ロロ体20 は 3-クロロ体 16 と同様にマウス代謝安定性の大幅な低下を示した。一方で、非
常に興味深いことに、3-フルオロ-5-トリフルオロメチル体 3 は本誘導体の中で最も強力な
ヒト11β-HSD1 阻害活性を示し(IC50 = 4.8 nM)、良好なマウス肝ミクロソーム安定性を示
した(mouse CLint = 578 mL/min/kg)。3、5、18 の評価結果より、チオフェン環 3 位の置換
基に関しては、ヒト11β-HSD1 阻害活性とマウス肝ミクロソーム安定性の両立ためにはフ ルオロ基が最も適切であると示唆された。さらに、3、4 の評価結果より、チオフェン環 5 位におけるトリフルオロメチル基の導入がヒト11β-HSD1 阻害活性とマウス肝ミクロソー ム安定性の両立に最も有効であることが示された。なお、いずれの化合物もヒト 11β-HSD2 阻害活性は示さなかった。以上の結果より、3-フルオロ-5-トリフルオロメチル基が チオフェン環の置換基として最も適切であることが示された。
27
Table 5. Conversion of substituent on the thiophene ring
3–5, 14–16, 18–23 4 5 3 Compound R Human 11β-HSD1 IC50 (nM) Human 11β-HSD2 IC50 (nM) Mouse CLint (mL/min/kg) CLogP 2 H 23 >10000 1122 2.95 14a 5-F 11 >3000 1461 3.00 19 5-Cl 15 >10000 1494 3.34 21 5-Br 7.9 >3000 1445 3.54 22 5-Me 19 >10000 1983 3.06 23 5-CN 95 >30000 385 2.49 4 5-CF3 24 >10000 423 3.59 15 4-Br 12 >10000 1674 3.50 5 3-F 12 >10000 1666 2.90 16 3-Cl 14 >10000 6791 3.34 18 3-F, 5-Cl 8.7 >3000 2116 3.06 20 3,5-diCl 13 >10000 >9066 3.71 3a 3-F, 5-CF 3 4.8 >3000 578b 3.53 a) Hydrochloride salt. b) Evaluated with free form.
28 第四節 トリアゾール環4 位の構造活性相関 最後に、脂溶性及び嵩高さの増大によるヒト11β-HSD1 阻害活性の向上を目的とし、ト リアゾール環4 位の置換基変換を行った場合の評価結果を Table 6 に示す。構造活性相関 研究の効率化のため、トリアゾール環4 位の変換は 3-フルオロ-5-トリフルオロメチルチ オフェン誘導体よりも合成が容易な5-ブロモチオフェン誘導体で実施した。比較のために 化合物21 のデータを Table 6 に示した。シクロプロピル基をメチル基で置換した化合物 12 は、21 と比較してヒト 11β-HSD1 阻害活性は約 3 分の 1 に減弱した。本結果により、 トリアゾール環4 位にはある程度嵩高い置換基が必要であることが示唆された。アルキル 誘導体13, 26–28 は 21 と同等のヒト 11β-HSD1 阻害活性を示したが、26–28 はマウス肝ミ クロソーム安定性の低下を示した。また、ベンジル体29、フェネチル体 30 は 21 と比較 してヒト11β-HSD1 阻害活性の減弱を示した。本結果は、トリアゾール環 4 位はベンゼン 環を有する炭化水素基よりも、それを有さないアルキル基が適している可能性があること を示している。なお、評価した化合物においてはヒト11β-HSD2 阻害活性を示す化合物は 無かった。これらの結果に基づいて、シクロプロピル基がトリアゾール環4 位における最 も適切な置換基であると結論付けた。
29
Table 6. Conversion of the 4-position substituent of 1,2,4-triazole
12, 13, 26–30 4 Compound R Human 11β-HSD1 IC50 (nM) Human 11β-HSD2 IC50 (nM) Mouse CLint (mL/min/kg) CLogP 21 c-Pr 7.9 >3000 1445 3.54 12a Me 26 NTb NTb 3.30 13a Et 9.6 >3000 1298 3.62 26a n-Pr 8.6 >3000 2771 4.12 27a i-Pr 14 >10000 5746 4.07 28 cyclopropyl methyl 12 >10000 4190 4.03 29a Bn 22 NTb NTb 5.16 30 phenethyl 43 >10000 NTb 5.39 b) Hydrochloride salt. c) Not tested.
30 第五節 第二章のまとめ 著者は、ヒト 11β-HSD1 阻害活性の向上、マウス肝ミクロソーム安定性の改善を目指し てリード化合物2 の類縁体を合成し、それらのヒト 11β-HSD1 阻害活性、ヒト 11β-HSD2 阻 害活性、マウス肝ミクロソーム安定性を評価した。 最初に、脂溶性の低減を目的としてチオフェン環を各種含窒素ヘテロ芳香環へ変換した 類縁体について評価した。ピリジン体31、32 はヒト 11β-HSD1 阻害活性は減弱し、ピラジ ン体 17 はヒト 11β-HSD1 阻害活性が大幅に減弱した。また、ピロール体 24 はマウス肝ミ クロソーム安定性は改善するものの、ヒト 11β-HSD1 阻害活性は約 6 分の 1 に減弱した。 これらの結果は、末端チオフェン環の含窒素ヘテロ芳香環への変換は許容性が低いことを 示唆した。 次に、脂溶性及び嵩高さの増大によるヒト 11β-HSD1 阻害活性の向上を目的として合成 したトリアゾール環3 位変換体について評価した。化合物 25 はヒト 11β-HSD1 阻害活性が 消失し、トリアゾール環 3 位には置換基が必要であることを明らかにした。tert-ブチル体 8、シクロブチル体 9、シクロペンチル体 10、シクロへキシル体 11 はヒト 11β-HSD1 阻害 活性が若干向上し、嵩高い置換基が好ましいことが示唆された。しかしながら、脂溶性の 増大に伴いマウス肝ミクロソーム安定性が低下し、トリアゾール環 3 位の嵩高いアルキル 基への変換では、ヒト 11β-HSD1 阻害活性の向上とマウス肝ミクロソーム安定性の改善を 両立させることは困難であることを明らかにした。 続いて、チオフェン環の酸化代謝の抑制を目的としてチオフェン環に電子吸引性基等の
31 置換基を導入した類縁体について評価した。チオフェン環 5 位をハロゲン基に置換した、 フルオロ体 14、クロロ体 19、ブロモ体 21 は 2 と比較してヒト 11β-HSD1 阻害活性は向上 したが、マウス肝ミクロソーム安定性を改善する効果は得られなかった。一方で、トリフ ルオロメチル体 4 は 2 と同等のヒト 11β-HSD1 阻害活性を示し、マウス肝ミクロソーム安 定性は約 3 倍向上した。マウス代謝安定性の改善はチオフェン環の酸化的代謝の抑制によ るものであると推察された。次に、3,5-二置換誘導体について評価した。非常に興味深いこ とに、3-フルオロ-5-トリフルオロメチル体 3 が最も強力なヒト 11β-HSD1 阻害活性を示し (IC50 = 4.8 nM)、良好なマウス肝ミクロソーム安定性を示した(mouse CLint = 578 mL/min/kg)。以上の結果より、3-フルオロ-5-トリフルオロメチル基がチオフェン環の置換 基として最も良好であることを明らかにした。 最後に、脂溶性及び嵩高さの増大によるヒト11β-HSD1 阻害活性の向上を目的としてト リアゾール環4 位の置換基変換を行った類縁体について評価した。メチル基に変換した化 合物12 はヒト 11β-HSD1 阻害活性が減弱し、ある程度嵩高い置換基が必要であることが 示された。アルキル誘導体26–28 は 21 と同等のヒト 11β-HSD1 阻害活性を示したが、マ ウス肝ミクロソーム安定性の低下を示した。また、ベンジル体29、フェネチル体 30 はヒ ト11β-HSD1 阻害活性の減弱を示し、ベンゼン環を有する炭化水素基よりもベンゼン環を 有さないアルキル基が適している可能性があることを示した。トリアゾール環4 位への脂 溶性の増大、または嵩高い置換基の導入ではヒト11β-HSD1 阻害活性の向上とマウス肝ミ クロソーム安定性の改善を両立させることは困難であると判断し、シクロプロピル基がト リアゾール環4 位における最も適切な置換基であると結論付けた。
32 第三章 化合物 3 の抗糖尿病作用の評価、及びヒト 11β-HSD1 阻害活性、種差に関する 考察 第一節 化合物3 の in vivo 評価 前述のチオフェン誘導体の中で、最も強力なヒト 11β-HSD1 阻害活性及び良好なマウス 肝ミクロソーム安定性を示した化合物3 を in vivo 評価化合物として選択した。まず、3 の in vitro 評価結果を Table 7 に示した。また、3 との比較のために 2 のマウス 11β-HSD1 阻害 活性についても同時に評価した。マウス 11β-HSD1 阻害活性については、大腸菌に発現さ
せたマウスの組み換え11β-HSD1 タンパク(residues 24–292 with His-tag at N-terminal)を使用
し、HTRF®を用いて測定した。ヒト代謝安定性はヒト肝ミクロソームを用いて測定した。 3 はマウス 11β-HSD1 に対して中程度の阻害活性しか示さなかったが、強力なヒト 11β-HSD1 阻害活性を有し、ヒト肝ミクロソームに対しても代謝的に安定であった。続いて、経口投 与における3 の抗糖尿病作用を評価するために in vivo 試験を実施した(Figure 6)。7 週齢 雄性ob/ob マウスに対して、3 を 0.3、1、3、10 mg/kg の用量で 1 日 2 回 4 週間反復経口投 与し、最終投与の 12 時間後に血漿及び後腹膜脂肪組織を採取し、血糖値、インスリン値、 脂肪組織のコルチコステロン濃度を測定した。その結果、3 は用量依存的に血糖値、インス リン値を低下させた。3 はインスリン値を増加させることなく血糖値を低下させたことか ら、インスリン抵抗性改善作用を示したと考えている。また、3 は脂肪組織中でのコルチコ ステロンレベルの低下も示した。本結果より、3 は in vitro ではマウス 11β-HSD1 に対して
33 中程度の阻害活性しか示さなかったが、マウス代謝安定性が良好であることから体内動態 は良好であると推測しており、3 のマウス 11β-HSD1 阻害活性は脂肪組織中のコルチコステ ロンレベルを低下させるのに十分であり、11β-HSD1 阻害活性を介して in vivo において血 糖値、インスリン値の低下をもたらしたと考察している。さらに、3 はヒト 11β-HSD1 阻害 活性がマウス11β-HSD1 阻害活性に比べて約 40 倍強いことから、ヒトでの強い抗糖尿病作 用が期待される。
Table 7. Profiles of compound 3
Compound Human 11β-HSD1 IC50 (nM) Mouse 11β-HSD1 IC50 (nM) Human 11β-HSD2 IC50 (nM) Human CLint (mL/min/kg) Mouse CLint (mL/min/kg) 2 23 20 >10000 NTb 1122 3a 4.8 189 >3000 27c 578c a) Hydrochloride salt. b) Not tested.
34
Figure 6. Effects of repeated administration of 3 in male ob/ob mice (0.3–10 mg/kg, twice daily for
4 weeks, n=7–8). Values are presented as the mean ± SE. *p <0.05, **p <0.01 versus vehicle-treated group, using the Dunnett's multiple comparisons test.
35
第二節 化合物3 のヒト 11β-HSD1 阻害活性及び種差に関する考察
化合物3が非常に強力なヒト11β-HSD1阻害活性を有する理由を明らかにするために、3
とヒト
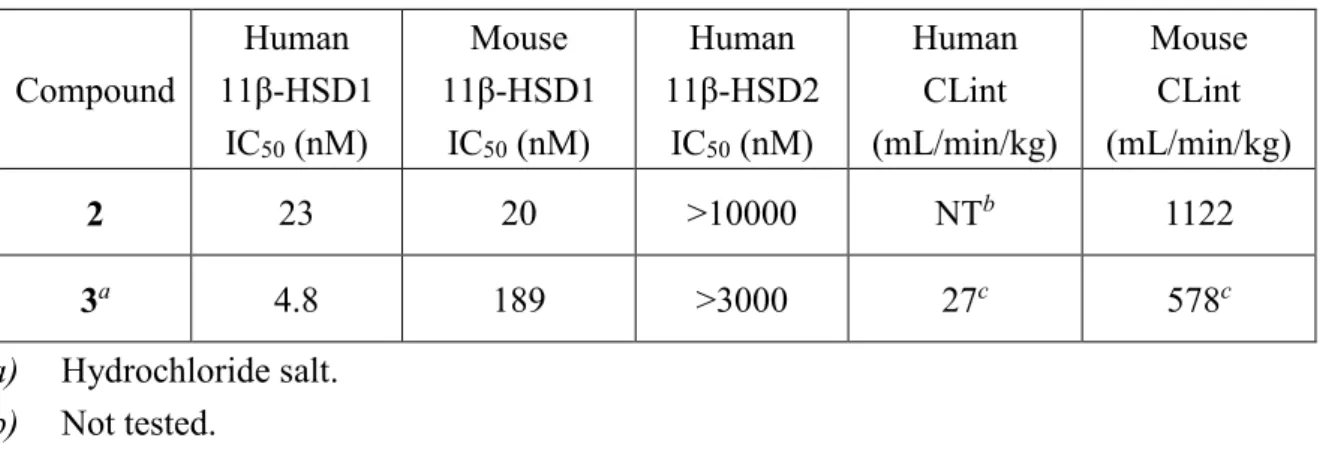
11β-HSD1タンパクとの相互作用に関するドッキングスタディを実施した。ヒト11β-HSD1モデルはトリアゾール型阻害剤とヒト11β-HSD1の複合体の結晶構造(PDB code 3D5Q)18)に基づいて作成した。3をFigure 7に示すようにGOLD version 5.5.1 19)を用いて
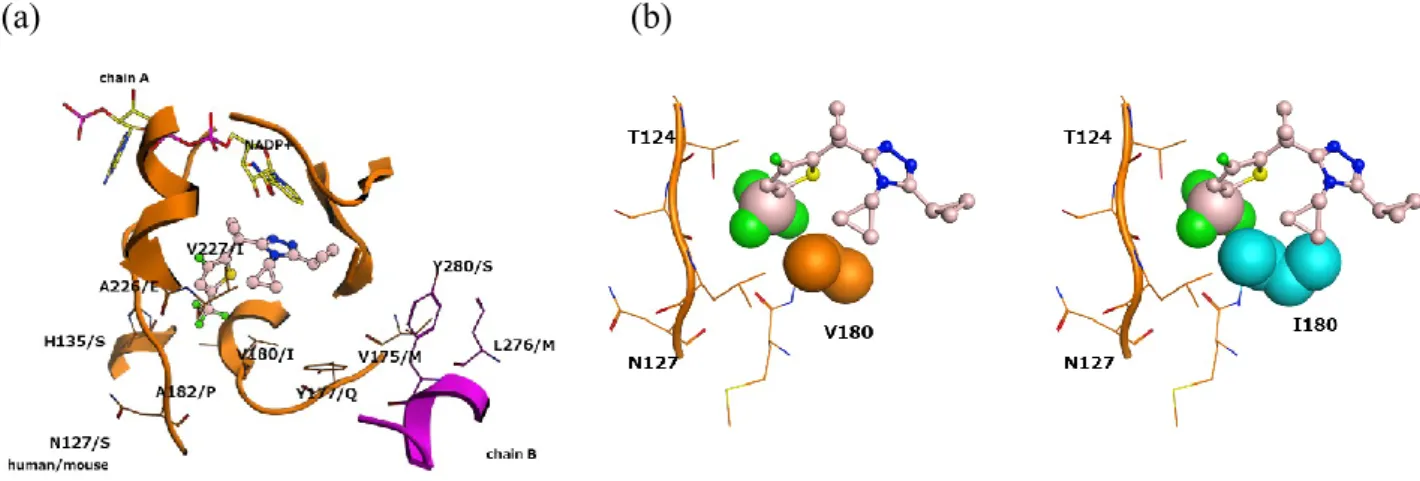
ドッキングした。その結果、3がステロイド結合部位に位置し、トリアゾール環1位の窒素 原子がTyr183、トリアゾール環2位の窒素原子がSer170とそれぞれ相互作用を有すること が示唆された(Figure 7a)。さらに、このモデルにおいては、3のチオフェン部位がヒト 11β-HSD1の疎水性ポケットに適合することが示された(Figure 7b)。これらの結果によ り、3がヒト11β-HSD1に対して強力な阻害活性を示したと考えている。 次に、3のヒトとマウスの11β-HSD1阻害活性の種差を分析するために、まずヒトとマウ スの11β-HSD1の配列相同性を調べた。その結果、ヒトとマウスの間で11β-HSD1のリガン ド結合部位周辺のいくつかのアミノ酸残基に種差があることを見出した(Figure 8a)。次 に、3とヒト11β-HSD1との間の相互作用のドッキングスタディにより、3のトリフルオロ メチル基がVal180を含む疎水性ポケットに適合することが示唆された。一方で、マウスに おける残基はバリンではなくイソロイシンであり、Figure 8bに示すようにバリンよりも立 体的に嵩高い。従って、3のトリフルオロメチル基は立体障害のためにこの位置に存在す ることが難しくなるため、3のマウス11β-HSD1阻害活性はヒト11β-HSD1阻害活性と比較し て減弱を示したと推察している。
36 (a) (b)
Figure 7. (a) Molecular modeling of the interaction between 3 (as a free base) and human
11β-HSD1. Best docking solution (lowest binding energy) was calculated using GOLD version 5.5.119)
for compound 3 (ball and stick diagram: blue is nitrogen, yellow is sulfur, green is fluorine and pale pink is carbon) when surrounded by human 11β-HSD1 active site residues. (b) Two-dimensional diagram prepared using the ligand interactions application in MOE.20)
(a) (b)
Figure 8. (a) Molecular modeling of the interaction between compound 3 (ball and stick diagram:
blue is nitrogen, yellow is sulfur, green is fluorine and pale pink is carbon) and human 11β-HSD1. The depicted residues are those that differ between human and mouse 11β-HSD1. For example, Val180 in human, Ile in mouse. (b) Simplified view of (a) focusing on the difference between human Val180 and mouse Ile180. Atoms relevant for the discrepancy in interactions are shown in Space-filling spheres. Mouse Ile180 is colored cyan.
37 第三節 第三章のまとめ 著者は、第二章で評価した誘導体の中で、最も強力なヒト 11β-HSD1 阻害活性、及び良 好なマウス肝ミクロソーム安定性を示した化合物 3 を選択し、経口投与における 3 の抗糖 尿病作用を評価するためにin vivo 試験を実施した。7 週齢雄性 ob/ob マウスに対して、3 を 0.3、1、3、10 mg/kg の用量で 1 日 2 回 4 週間反復経口投与した。その結果、3 は用量依存 的に血糖値、インスリン値の低下作用を示した。3 はインスリン値を増加させることなく 血糖値を低下させたことから、インスリン抵抗性改善作用を示したと考えている。また、3 は脂肪組織中でのコルチコステロンレベルの低下も示したことから、11β-HSD1 阻害作用を 介してコルチコステロンレベルを低下させin vivo において血糖値、インスリン値の低下を もたらしたと考察した。 3が非常に強力なヒト11β-HSD1阻害活性を有する理由を明らかにするために、3とヒト 11β-HSD1タンパクとの相互作用に関するドッキングスタディを実施した。その結果、3が ステロイド結合部位に位置し、トリアゾール環1位の窒素原子がTyr183、トリアゾール2位 の窒素原子がSer170とそれぞれ相互作用を有することが示唆された。さらに、3のチオ フェン部位がヒト11β-HSD1の疎水性ポケットに適合することが示された。これらの結果 により、3がヒト11β-HSD1に対して強力な阻害活性を示したと考えている。 次に、3のヒトとマウスの11β-HSD1阻害活性の種差を理解するためにヒトとマウスの 11β-HSD1の配列相同性を調べた。その結果、ヒトとマウスの間で11β-HSD1のリガンド結 合部位周辺のいくつかのアミノ酸残基に種差があることを見出した。また、3のトリフル
38
オロメチル基がVal180を含む疎水性ポケットに適合することが示唆された。一方で、マウ
スにおける残基はバリンではなくイソロイシンであり、3のトリフルオロメチル基は立体
障害のためにこの位置に存在することが難しくなるため、マウス11β-HSD1阻害活性はヒ
39 第四章 化合物3の合成法の改良 第一節 化合物3の合成① 化合物3の種々の高次評価、毒性試験を実施するためには3の大量供給が必要である。 従って、安価な原料からの合成が必要と考え合成ルートの検討を行った。その結果を Scheme 10に示す。 安価な原料である化合物59を出発原料とし、59のチオフェン環3位のフッ素化、生成し たフルオロカルボン酸60のCDIによる活性化、NaBH4による還元を経てアルコール体61を 合成した。61のヒドロキシ基を塩化チオニルを用いて塩素に変換して62とし、塩素をシア ノ基に置換することにより化合物39を得た。次に、ニトリル39のα位をジメチル化して40 とした後、KOHによるシアノ基の加水分解により得られたカルボン酸41をメチル化する ことでエステル63を得た。続いて、NISを用いて63のチオフェン環5位をヨウ素化し、生成 した63のヨウ素をsodium trifluoroacetate、ヨウ化銅を用いてトリフルオロメチル基に置換 することで低収率ながらトリフルオロメチル体34を得た。34をヒドラジン一水和物と加熱 することでヒドラジド35に変換し、35と36をMeOTfを用いて環化させることでトリアゾー ル3’を得た。最後に、造塩反応を行って、化合物3を12工程、総収率0.2%で合成した。59 から3の合成は達成したが、工程数が長い、フッ素化の収率が低い、トリフルオロメチル 化の収率が低いという3つの問題があり、スケールアップを実施するためにはさらなる改 善が必要と考え、上記3点の課題解決を検討することとした。
40 12工程、総収率0.2% 問題点 ①工程数が長い ②59→60、フッ素化(収率31%) ③64→34、トリフルオロメチル化(収率6%) 3' 36 41% 2 steps 59 l 60 95% HCl 84% 92% 61 g 6% 62 89% 42% d 35 95% 64 90% 34 98% c 31% k 63 i j b h e f 39 a 40 41 3
Scheme 10. Synthetic route of 3: part 1. Reagents and conditions: (a) n-BuLi, THF, –78°C, 1h then
N-fluorobenzenesulfonimide, THF, –78°C, 5 h, r.t., overnight, 31%; (b) CDI, THF, r.t., 1 h then NaBH4, H2O, r.t., 3 h, 42%; (c) SOCl2, cat. pyridine, CH2Cl2, r.t., 6 h, 92%; (d) NaCN, DMSO, r.t.,
2 h, 84%; (e) MeI, NaH, DMF, r.t., 20 min, 90%; (f) KOH, ethylene glycol, 190°C, 2.5 h, 98%; (g) MeI, K2CO3, DMF, r.t., 15 h, 95%; (h) NIS, AcOH, CH2Cl2, r.t., 2 d, 95%; (i) sodium
trifluoroacetate, CuI, NMP, 180°C, 5 h, 6%; (j) hydrazine monohydrate, EtOH, 80°C, 24 h, 89%; (k) 36, MeOTf, 60°C, 30 min then Et3N, toluene, 60°C, 24 h, 90°C, 9 h, 110°C, 15 h; (l) 4M HCl/EtOAc, EtOAc, 41%, 2 steps.
41 第二節 化合物3の合成② 著者らは前述の3つの課題を解決するために、まずは原料を変更することによる工程数 の短縮を検討した。その結果をScheme 11に示す。 化合物59以外に入手可能な安価な原料として化合物65を選択した。エステル65のα位を ジメチル化し、得られた化合物66のチオフェン環5位をNISを用いてヨウ素化して化合物67 を合成した。次に、67のヨウ素をsodium trifluoroacetate、ヨウ化銅を用いてトリフルオロ メチル基に置換し、得られた化合物37のエステル基の加水分解によりカルボン酸68を合成 した。68のチオフェン環3位のリチオ化、続くN-fluorobenzenesulfonimideによるフッ素化を 行ってカルボン酸誘導体33を合成し、33のエステル化を経てメチルエステル体34を合成し た。続いて34のエステル基をEtOH溶媒中、ヒドラジン一水和物と加熱することでヒドラ ジド35を合成し、35と36をMeOTfを用いて環化させてトリアゾール3’を得た。最後に、造 塩反応を行って、3を9工程、総収率4.7%で合成した。本検討では工程数を3工程短縮し総 収率を20倍以上改善したが、スケールアップのためには総収率のさらなる改善が必要であ る。総収率が低い原因はトリフルオロメチル化とフッ素化であるため、両反応の収率改善 に絞って検討を行うこととした。
42 67 3' 37 68 33 66 9工程、総収率4.7% 問題点 ①67→37、トリフルオロメチル化(収率44%) ②68→33、フッ素化(収率38%) 65 99% 94% 38% 84% 44% d b c f 41% 2 steps e HCl i 89% 35 34 99% h g a 3
Scheme 11. Synthetic route of 3: part 2. Reagents and conditions: (a) MeI, NaH, NMP, <15°C, 1 h,
99%; (b) NIS, AcOH, r.t., 2 h, 99%; (c) sodium trifluoroacetate, copper iodide, NMP, 180°C, 5 h, 44%; (d) 1 M NaOH aq., EtOH, r.t., 15 h, 94%; (e) n-BuLi, THF, –50 to –45°C, 30 min then N-fluorobenzenesulfonimide, THF, –78°C, 1 h, r.t., 15 h, 38%; (f) MeI, K2CO3, DMF, r.t., 15 h, 84%;
(g) hydrazine monohydrate, EtOH, 80°C, 24 h, 89%; (h) 36, MeOTf, 60°C, 30 min then Et3N, toluene, 60°C, 24 h, 90°C, 9 h, 110°C, 15 h; (i) 4 M HCl/EtOAc, EtOAc, 41%, 2 steps.
トリフルオロメチル化の検討結果をTable 8に示す。Scheme 11の反応条件であるentry 1 では、中程度の収率(44%)、及びsodium trifluoroacetateを10当量、ヨウ化銅を5当量用い ること、反応温度が高いことによる大量合成の際の操作性の悪さが課題であった。本問題 を解決するためにトリフルオロメチル化試薬をmethyl 2-(fluorosulfonyl)difluoroacetateに変 更することで91%の収率で化合物37を合成することができた(entry 2)。また、試薬の当 量の削減、反応温度の低下にも成功し、反応のスケールアップの際の操作性も向上した。 収率が改善した理由は、試薬の変更によりヨウ素が還元除去された副生成物の生成が抑制 されたためであると推察している。
43
Table 8. トリフルオロメチル化の検討
67 37
entry reagents solvent temp. (°C) yield (%) 1 CF3CO2Na (10 eq.) CuI (5 eq.) NMP 180 44 2 FSO2CF2CO2Me (3 eq.) CuI (0.3 eq.) DMF 95 91 続いて、フッ素化の検討結果をTable 9に示す。種々の検討の結果、反応試薬であるN- fluorobenzenesulfonimideに問題があると推察し試薬の前処理を検討した。その結果、N-fluorobenzenesulfonimideをジクロロメタンに溶解し、無水硫酸ナトリウムで乾燥後、不溶 物を濾別後に反応に用いることでScheme 9の条件であるentry 1と比較して収率が改善し収 率68%で化合物33を合成することに成功した(entry 2)。反応試薬であるN-fluorobenzenesulfonimideに水が含まれていたか、もしくは含まれていた何らかの不純物を 前処理で除去することができたことで、収率が改善したと推察している。 Table 9. フッ素化の検討 68 33 entry reagents リチオ化 条件 フッ素化 条件 (PhSO2)2NFの前処理 yield (%) 1 n-BuLi (2.5 eq.) (PhSO2)2NF (1.6 eq.) -50~-45°C 30 min -78°C, 1 h, r.t., 15 h 無し 38 2 n-BuLi (2.5 eq.) (PhSO2)2NF (1.5 eq.) -65~-55°C 30 min -78°C, 1 h, 0°C, 2 h CH2Cl2に溶解後、無 水硫酸ナトリウムで 乾燥、不溶物をろ過 68
44 化合物3の合成ルートの最終的な検討結果をScheme 12に示す。著者は原料の変更による 工程数の短縮、反応試薬の変更によるトリフルオロメチル化の収率向上、反応試薬の前処 理によるフッ素化の収率向上を達成することにより、9工程、総収率21%で3の合成に成功 した。本ルートにより3を32 g合成しており、さらなるスケールアップのための基礎が 整った。 67 80% 3' 37 68 33 66 9工程、総収率21% 本ルートで3を32 g合成 65 99% 94% 68% 84% 91% d b c f 61% e HCl i 88% 35 34 99% h g a 3
Scheme 12. Synthetic route of 3: part 3. Reagents and conditions: (a) MeI, NaH, NMP, <15°C, 1 h,
99%; (b) NIS, AcOH, r.t., 2 h, 99%; (c) methyl 2-(fluorosulfonyl)difluoroacetate, CuI, DMF, 95°C, 7 h, 91%; (d) 1 M NaOH aq., EtOH, r.t., 15 h, 94%; (e) n-BuLi, THF, –65 to –55°C, 30 min then N-fluorobenzenesulfonimide, THF, –78°C, 1 h, 0°C, 2 h, 68%; (f) MeI, K2CO3, DMF, r.t., 15 h, 84%;
(g) hydrazine monohydrate, EtOH, 80°C, 15 h, 88%; (h) 36, MeOTf, 60°C, 30 min then DIPEA, toluene, 60°C, 15 h then DIPEA, 110°C, 15 h, 61%; (i) 4 M HCl/EtOAc, EtOAc, r.t., 30 min, 80%.
45 第三節 第四章のまとめ 著者は、化合物3の種々の高次評価、毒性試験実施の際に必要となる大量供給に向け て、合成ルートの検討を実施した。 まず、安価な原料である化合物59を出発原料とした合成ルートの検討を行い、3を12工 程、総収率0.2%で合成した(Scheme 10)。3の合成は達成したが、工程数が長い(12工 程)、フッ素化の収率が低い(31%)、トリフルオロメチル化の収率が低い(6%)とい う3つの問題により総収率は非常に低く(0.2%)、スケールアップを実施するためにはさ らなる改善が必要と考えた。 次に、化合物65を出発原料とし合成ルートの検討を行った(Scheme 11)。その結果、3 を9工程、総収率4.7%で合成することができた。本検討では工程数を3工程短縮し、総収率 を20倍以上改善したが総収率のさらなる改善が必要であった。総収率が低い原因はトリフ ルオロメチル化とフッ素化の低収率(それぞれ、44%及び38%)にあるため、両反応の収 率改善を目指して検討を行った。トリフルオロメチル化では、トリフルオロメチル化試薬 をmethyl 2-(fluorosulfonyl)difluoroacetateに変更することで、91%の収率で化合物37を合成す ることができた(Table 8)。また、フッ素化の検討では、N-fluorobenzenesulfonimideを前 処理することで、収率68%で化合物2を合成することに成功した(Table 9)。 以上のように、原料の変更による工程数の短縮、反応試薬の変更によるトリフルオロメ チル化の収率向上、反応試薬の前処理によるフッ素化の収率向上に成功し、9工程、総収 率21%で3の合成を達成した(Scheme 12)。
46 総括 糖尿病とは、膵臓のβ細胞からのインスリン分泌不全、または抹消組織におけるインス リン抵抗性に起因するインスリン作用不足の結果として、高血糖が慢性的に持続する代謝 疾患であり、治療が遅れると糖尿病合併症を発症する。従って、早期の診断、治療が必要 である。しかしながら、限られた薬理学的効果や有害な副作用のために、多くの 2 型糖尿 病患者では十分な血糖コントロールは達成されておらず、新たな作用機序を有する薬剤の 開発が求められている。11-HSD1 は肝臓や脂肪組織において不活性型グルココルチコイド を活性型グルココルチコイドに変換する酵素である。11β-HSD1 阻害剤は、不活性型グルコ コルチコイドから活性型グルココルチコイドへの変換を抑制することにより、活性型グル ココルチコイドによる、高血糖、インスリン抵抗性および高脂血症などの代謝異常を改善 すると期待される。著者は、糖尿病治療薬としての新規11β-HSD1 阻害剤の創製を目指し、 トリアゾール誘導体の構造活性相関研究を行った。 第一章では、リード化合物 2 のヒト 11β-HSD1 阻害活性の向上、マウス肝ミクロソーム 安定性の改善の 2 点の課題解決を目指して、構造活性相関研究に必要な 2 の類縁体の合成 を行った(Figure 9)。ヒト 11β-HSD1 阻害活性の向上に関しては、トリアゾール環 3、4 位 の脂溶性の増大や嵩高い置換基の導入を中心に構造変換を行った。一方で、マウス代謝安 定性の改善に関しては、チオフェン環を含窒素ヘテロ環に変換することによる脂溶性の低 減、及びチオフェン環の酸化代謝を抑制するために、電子吸引性置換基を中心にチオフェ
47 ン環への置換基導入を行った。 例) 例) ・脂溶性の増大 ・嵩高い置換基の導入 例) 3 例) 4 ・脂溶性の増大 ・嵩高い置換基の導入 ・含窒素ヘテロ環の導入による脂 溶性の低下 ・置換基導入によるチオフェンの酸化 代謝のブロック 青:ヒト11β-HSD1阻害活性向上を指向 赤:マウス肝ミクロソーム安定性改善を指向 human 11β-HSD1: IC50 = 23 nM mouse CLint = 1122 mL/min/kg
2 Figure 9. 2からの構造変換 第二章では、ヒト 11β-HSD1 阻害活性の向上、マウス肝ミクロソーム安定性の改善を目 指し、第一章で合成した 2 の各種類縁体について構造活性相関を考察した。最初に、脂溶 性の低減を目的としてチオフェン環を各種含窒素ヘテロ芳香環へ変換した類縁体の評価を 行い、含窒素ヘテロ芳香環への変換は許容性が低いことを明らかにした(Table 3)。次に、 ヒト 11β-HSD1 阻害活性の向上を目的として合成したトリアゾール環 3 位変換体の評価を 行い(Table 4)、嵩高い置換基が好ましいことを明らかにした。一方で、脂溶性の増大に伴 いマウス肝ミクロソーム安定性が低下し、嵩高いアルキル基への変換では、ヒト11β-HSD1 阻害活性の向上とマウス肝ミクロソーム安定性の改善を両立させることは困難であること を明らかにした。続いて、チオフェン環の酸化代謝を抑制するために、チオフェン環へ電 子吸引性基等の置換基を導入した類縁体を評価し(Tabele 5)、3-フルオロ-5-トリフルオロ メチル体3 が最も強力なヒト 11β-HSD1 阻害活性を示し(IC50 = 4.8 nM)、良好なマウス肝
48
ミクロソーム安定性を示したことから(mouse CLint = 578 mL/min/kg)、3-フルオロ-5-トリ フルオロメチル基がチオフェン環の置換基として最も良好であることを明らかにした。最 後に、ヒト 11β-HSD1 阻害活性の向上を目的として合成したトリアゾール環 4 位変換体の 評価を実施し(Table 6)、ベンゼン環を有する置換基よりもそれを有さないアルキル基が適 している可能性があることを示した。一方で、脂溶性の増大、または嵩高い置換基の導入 では、ヒト 11β-HSD1 阻害活性の向上とマウス肝ミクロソーム安定性の改善を両立させる ことは困難であることを明らかにし、シクロプロピル基がトリアゾール環 4 位における最 も適切な置換基であると結論付けた。以上の結果から、3 を in vivo 評価化合物として選択 した。 HCl 3 第三章では、最も強力なヒト 11β-HSD1 阻害活性、及び良好なマウス肝ミクロソーム安 定性を示した 3 の糖尿病モデルマウスでの評価とヒト 11β-HSD1 阻害活性、ヒトとマウス の 11β-HSD1 阻害活性の種差に関する考察を行った。最初に、経口投与における 3 の抗糖 尿病作用を評価した(Figure 6)。7 週齢雄性 ob/ob マウスに対する経口投与の結果、3 は用 量依存的に血糖値、インスリン値の低下作用を示し、インスリン抵抗性改善作用を示した と考察した。次に、3 が非常に強力なヒト 11β-HSD1 阻害活性を有する理由を明らかにする ために、3 とヒト 11β-HSD1 タンパクとの相互作用に関するドッキングスタディを行い
49 (Figure 7)、3 のトリアゾール環 1 位の窒素原子が Tyr183、トリアゾール環 2 位の窒素原 子が Ser170 とそれぞれ相互作用を有し、3 のチオフェン部位がヒト 11β-HSD1 の疎水性ポ ケットに適合することを示した。これらの結果、3 がヒト 11β-HSD1 に対して強力な阻害活 性を示したと考察した。最後に、3 のヒトとマウス間の 11β-HSD1 阻害活性の種差を理解す るためにヒトとマウスの 11β-HSD1 の配列相同性を調べたところ(Figure 8)、ヒトとマウ スの間で 11β-HSD1 のリガンド結合部位周辺のいくつかのアミノ酸残基に種差があること を見出した。また、ヒトでは3 のトリフルオロメチル基が Val180 を含む疎水性ポケットに 適合することが示唆されたが、マウスにおける残基はバリンではなくイソロイシンであり、 3 のトリフルオロメチル基は立体障害のためにこの位置に存在することが難しくなるため、 マウス11β-HSD1 阻害活性はヒト 11β-HSD1 阻害活性と比較して減弱を示したと考察した。 第四章では、3の合成ルートの検討を行った。最初に、安価な原料である化合物59を出 発原料とした合成ルートの検討を行い、3を12工程、総収率0.2%で合成した(Scheme 10)。次に、工程数が長い、フッ素化、トリフルオロメチル化が低収率という3つの課題 解決のために合成経路の検討を行った。まず、化合物65を出発原料として合成ルートを再 検討することで、工程数を3工程短縮し総収率を20倍以上改善した(Scheme 11)。次に、 第2の経路でも問題として残されたトリフルオロメチル化(収率44%)とフッ素化(収率 38%)の工程について、さらに詳細な検討を行った。トリフルオロメチル化の検討ではト リフルオロメチル化試薬を変更することで91%の収率で化合物37を合成し(Table 8)、 フッ素化の検討ではフッ素化試薬を前処理することで、収率68%で化合物33を合成するこ
50 とに成功した(Table 9)。これらの検討の結果、原料の変更による工程数の短縮、反応試 薬の変更によるトリフルオロメチル化の収率向上、反応試薬の前処理によるフッ素化の収 率向上に成功し、9工程、総収率21%で3の合成を達成した(Scheme 12)。 以上、糖尿病治療薬としての新規 11β-HSD1 阻害剤の創製を目指し、トリアゾール誘導 体の構造活性相関研究を行った結果、強力なヒト 11β-HSD1 阻害活性を有し、in vivo で血 糖低下作用、インスリン低下作用を示す 3 を見出した。本研究成果が今後の糖尿病治療薬 の研究開発、及び11β-HSD1 阻害剤の研究開発に貢献することを期待する。
51 実験の部
Chemistry
Starting materials and reagents are commercially available. 1H NMR spectra were recorded on
Varian 300-MR, Varian 400-MR, Varian VNS-400 or JEOL LAMBDA and chemical shifts were expressed in δ (ppm) values with trimethylsilane as an internal reference (s = singlet, d = doublet, dd = double doublet, ddd = double double doublet, t = triplet, q = quartet, m = multiplet, br = broad and brs = broad singlet). Mass spectra (MS) were recorded on JEOL JMS-LX2000, Waters ZQ2000, Thermo Electron TRACE DSQ, Thermo Electron LCQ Advantage or Thermo Scientific Exactive Plus. Elemental analyses were performed with Elementar Vario EL III (C, H, N) and DIONEX ICS-5000 (S, halogen) instruments, and results were within ±0.4% of theoretical values. All reactions were carried out using commercially available reagents and solvents without further purification.
第一章 新規トリアゾール誘導体の合成
Methyl 2-[3-fluoro-5-(trifluoromethyl)thiophen-2-yl]-2-methylpropanoate (34)
Dipotassium carbonate (6.20 g, 44.9 mmol) and MeI (2.27 mL, 36.5 mmol) were added to an ice-cooled solution of 2-[3-fluoro-5-(trifluoromethyl)thiophen-2-yl]-2-methyl propanoic acid (33, 7.67g, 29.9 mmol, CAS No.; 950604-93-0) in DMF (30 mL), and the mixture was stirred at room temperature for 15 h. The reaction mixture was diluted with water (60 mL) and extracted with Et2O.
52
The organic layer was washed with water, dried over Na2SO4, filtered and concentrated in vacuo. The
resulting residue was purified by column chromatography on silica gel (hexane–Et2O) to give the title
compound 34 (6.81 g, 25.2 mmol, 84%) as a pale yellow syrup. 1H-NMR (400 MHz, CDCl
3) δ: 1.65
(6H, s), 3.73 (3H, s), 7.12–7.13 (1H, m).
2-[3-Fluoro-5-(trifluoromethyl)thiophen-2-yl]-2-methylpropanehydrazide (35)
Hydrazine monohydrate (1.0 mL, 21 mmol) was added to a solution of 34 (322 mg, 1.19 mmol) in EtOH (2 mL) and the mixture was stirred at 80°C for 24 h. The reaction mixture was concentrated in vacuo. The resulting residue was diluted with saturated aqueous sodium hydrogen carbonate solution and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered and
concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (CHCl3–MeOH) to give the title compound 35 (286 mg, 1.06 mmol, 89%) as a pale yellow syrup. 1H-NMR (400 MHz, CDCl
3) δ: 1.66 (6H, s), 2.10–3.90 (2H, br), 6.80–7.04 (1H, m), 7.17 (1H, s).
CI-MS m/z: 271 [M+H]+.
3,4-Dicyclopropyl-5-{2-[3-fluoro-5-(trifluoromethyl)thiophen-2-yl]propan-2-yl}-4H-1,2,4-triazole monohydrochloride (3)
A mixture of 36 (220 mg, 1.76 mmol, CAS No.; 1453-50-5) and MeOTf (200 μL, 1.77 mmol) was stirred at 60°C for 30 min and diluted with toluene (4 mL). A solution of Et3N (500 μL, 3.59