博士論文
Role of dopamine D2 receptor in the generation of nicotine
dependence
(ニコチン依存性形成におけるドパミン D2 受容体の役
割に関する研究)
令和元年度
東北大学大学院薬学研究科
生命薬科学専攻 薬理学分野
GOFARANA WILAR
i
LIST OF CONTENT
Page
List of Content..……….………...……i
List of Abbreviation..……….….………..………1
Chapter 1 General Introduction………..………..3
1.1 Nicotine……….…….………...3
1.2 Nicotinic Acetilcholine Receptors (nAChRs)……….……….…….5
Chapter 2 Inhibition of Nicotine Dependence by Curcuminoid is Associated with Reduced Acetylcholinesterase Activity in the Mouse Brain………..……….……..8
2.1 Introduction………...8
2 . 1 . 1 Acetylcholine………...8
2 . 1 . 2 Curcuminoids………..……..……….…...11
2.2 Objectives………..………12
2.3 Materials and Methods……….………...…….13
2 . 3 . 1 Animals and Materials………13
2 . 3 . 2 Drug administration………13
2 . 3 . 3 Conditioned Place Preference Test……….14
2 . 3 . 4 Habituation……….15
2 . 3 . 5 Pre-conditioning test………...………15
2 . 3 . 6 Conditioning and Post-conditioning testing………...15
2 . 3 . 7 Extinction and abstinence test………16
2 . 3 . 8 Priming nicotine/relapse……….16
2 . 3 . 9 CPP (Conditioned Place Preference) Apparatus………16
2.3.10 Acetylcholinesterase activity assay………17
2.3.10.1 Brain sample preparation………...17
2.3.10.2 Assay of enzyme activity………...17
2.3.11 Statistical analysis………..18
2.4 Results……….19
2 . 4 . 1 Nicotine administration generates nicotine dependence and relapse in mice…...19
2 . 4 . 2 Curcuminoid attenuates the nicotine-induced preference ratio in the CPP test at conditioning and priming phases………....20
2 . 4 . 3 Nicotine-induced Withdrawal is attenuated by pre -administration with curcuminoid……….21
ii
2 . 4 . 5 Curcuminoid inhibits acetylcholinesterase activity during nicotine reward behavior
………23
2.5 Discussion……….………25
Chapter 3 Crucial Role Of Dopamine D2 Receptor Signaling In Nicotine -Induced Conditioned Place Preference ………..…… 31
3.1 introduction…..……….31
3 . 1 . 1 Nicotine Reward System……….31
3 . 1 . 2 Nucleus Accumbens…………..……..………32
3 . 1 . 3 Hippocampus………...33
3 . 1 . 4 Dopamine Receptor………...……… ……….34
3 . 1 . 5 Dopamine Signal on Nicotine Dependence……….35
3.2 Objectives………37
3.3 Materials and Methods………38
3 . 3 . 1 Animals……….……….………38
3 . 3 . 2 Experimental design…………...………39
3. 3. 2 . 1 Induction of CPP by nicotine………39
3. 3. 2 . 2 D1 and D2 receptor antagonist alleviates nicotine-induced CPP…….39
3 . 3. 2 . 3 Nicotine CPP test for WT and D2RKO mice………...39
3 . 3. 2 . 4 Drug administration………..40
3 . 3. 2 . 5 CPP test……….41
3 . 3. 2 . 6 Acclimatization……….41
3 . 3. 2 . 7 Preconditioning test……….………..41
3 . 3. 2 . 8 Conditioning……….……….41
3 . 3 . 3 Locomotor activity test………...………42
3 . 3 . 4 Immunoblot analysis……….………42
3 . 3 . 5 Immunohistochemistry………..………...….43
3 . 3 . 6 Data Analysis and Statistical evaluation……….44
3.4 Results……….……….…45
3 . 4 . 1 Nicotine promotes CPP through D1 and D2 receptor activation………45
3 . 4 . 2 Nicotine-induced CPP was abolished in D2RKO mice………..47
3 . 4 . 3 Protein kinase signals are elevated after nicotine-induced CPP in NAc…..…….49
3 . 4 . 4 Protein kinase signals in the hippocampal CA1 are elevated after nicotine-induced CPP……….51 3 . 4 . 5 Immunohistochemical signals of CaMKII and ERK are elevated after
nicotine-iii
induced CPP in NAc………...…………54
3 . 4 . 6 Immunostaining signal ERK is enhanced after chronic nicotine exposure…..….56
3 . 4 . 7 Involvement of nAChR signal on activation of dopamine D2 receptor transmission……….57 3.5 Discussion………61 Chapter 4 Summary………72 Acknowledgement………..77 References……….………...79 List of Publications………..………...111
1
LIST OF ABBREVIATION
AChE = Acetyl cholinesterase Ach = Acetylcholine
AChEIs = Acetyl cholinesterase inhibitors7 AcS = Nucleus accumbens shell
ACC= Anterior Singulate Cortex ANOVA = Analysis of variant
α4β2nAChR = Alpha 4 Betha 2 Nicotinic Acetylcholine Receptors α7nAChR = Alpha 7 Nicotinic Acetylcholine Receptors
BDNF = Brain Derived Neurotropic Factor
CaMKII = Calcium/calmodulin-dependent Protein Kinase II CNS = Central Nervous System
CHRNA4 = nicotinic acetylcholine receptor (nAChR) α4 subunit CPP= Conditioned Placed Preference
CREB = cAMP Response Element Binding D2RKO = Dopamine D2 Receptor Knock Out DLS = Dorsalateral Striatum
DR = Dopamine Receptor D1R= Dopamine D1 Receptors D2R = Dopamine D2 Receptors
DTNB = 5,5-dithiobisnitro benzoic acid; ROS, Reactive oxygen species ERK = Extracellular Regulated Kinase
FAAH = Fatty acid amide hydrolase GABA = Gamma amino butyric acid
2 IL-1β = Interleukin 1 beta
JNK = Jun N-terminal kinases JNK LTP = Long Term Potentiation MSN = Medium Spiny neurons
MAPK = Mitogen activated protein kinase nAChRs = Nicotinic Acetylcholine Receptors NQ = Neonatal Quinpirole
NS = Neonatal Saline NAc = Nucleus Accumbens
NTRK2 = Neurotrophic Tyrosine Kinase Receptor 2 NSAIDs = Non-steroid anti-inflammatory drugs
pCREB = Phosphorylation cAMP Response Element Binding PKC= Phosphokinase C
PVDF = Polyvinylidene Difluoride PtC = Parietal Association Cortex
PPTg/LDTg = pedunculopontine/lateral dorsal tegmentum TNFα = Tumor necrosis factor alpha
VGCCs = Voltage-Gated Calcium Channels VTA = Ventral Tegmental Area
3
CHAPTER 1
GENERAL INTRODUCTION 1.1 Nicotine
Nicotine is an active compound and the main addictive material in tobacco products, nicotine dependence symptoms characterized by compulsive use, craving, tolerance from continued use and withdrawal upon cessation (Benowitz, 1999). Nicotine dependence is a chronic brain disorder and a worldwide primary public health issue. Tobacco smokers are motivated by nicotine dependence and this is the primary difficulty that prevents smoking cessation (Henningfield, Miyasato and Jasinski, 1985). Prolonged tobacco uses results in physiologic dependence and a behavioral compulsion to use tobacco. Nicotine establishes and maintains tobacco addiction by complex actions that affect the neurochemistry of the brain (Benowitz, 2010). Nicotine from cigarette smoke is rapidly absorbed in the lungs and then quickly passes into the brain in tens of second (Benowitz, Hukkanen and Jacob, 2009). In the bloodstream at pH 7.4, nicotine exists in ionized (69%) and unionized forms (31%), The unionized form can pass through lipid membranes and directly alter intracellular signaling (Rezvani et al., 2007).
Figure 1. (s) Nicotine ((S)-3-[1-Methylpyrrolidin-2-yl] pyridine)
The evidence on human showed the autopsy samples from smokers, the highest affinity for nicotine is in the liver, kidney, spleen, and lung and the lowest affinity in adipose tissue (Urakawa et al., 1994). In fatal case of intentional overdose of nicotine patches, brain nicotine levels reached 2-fold higher than in peripheral blood and about 40% of the nicotine
4
level in the liver (Kemp et al., 1997). Blood or plasma nicotine concentrations sampled in the afternoon on smokers generally range from 10 to 50 ng/ml (Benowitz, Hukkanen and Jacob, 2009). During daily smoking Typical trough concentrations are 10 to 37 ng/ml, and typical peak concentrations range between 19 and 50 ng/ml (Schneider et al., 2001). Nicotine is extensively metabolized to several of metabolites by the liver. Six primary metabolites of nicotine have been identified, the most important metabolite of nicotine in most mammalian species is the lactam derivative cotinine. In humans, around 70% to 80% of nicotine is converted to cotinine (Benowitz and Jacob, 1994). Nicotine is excreted by glomerular filtration and tubular secretion on kidney, with variable reabsorption affect by urinary pH. with uncontrolled urine pH, renal clearance averages 35 - 90 ml/min, accounting for the elimination of about 5% of total clearance. In acid urine, nicotine is mostly ionized, and tubular reabsorption is minimized; renal clearance 600 ml/min (urinary pH 4.4), depending on urinary flow rate (Benowitz and Jacob, 1985). Moreover, nicotine and cotinine is excreted in sweat of smokers but the total amount of nicotine excreted in sweat has not been quantitated in relation to urinary excretion (Hukkanen, Iii and Benowitz, 2005).
Nicotine binds to nicotinic acetylcholine receptors (nAChRs), which are pentamers composed of α2, α4 α7, α10, and β2-β4 subunits (Changeux, 2010b). nAChRs are widely distributed in the central nervous system (CNS), including cortical and limbic regions. These receptors are critical for drug addiction via stimulation of synaptic activity in the hippocampus, amygdala, ventral tegmental area (VTA), and nucleus accumbens (NAc) (Leslie, Mojica and Reynaga, 2013). Nicotine also increases the firing rate of midbrain dopamine neurons via stimulation of α4β2 nAChRs to promote nicotine dependence (De Biasi and Dani, 2011).
5
The α7 homo-oligomer and α4β2 hetero-oligomer are two major subtypes of nAChRs in the mammalian brain (Changeux, 2010b). Both receptors regulate nicotine dependence and chronic nicotine exposure selectively upregulates the density of α4β2, which enhances nicotine addiction in rats (Nguyen, Rasmussen and Perry, 2003), and increased α7 nAChR in the VTA in a nicotine dependence rat model (Nomikos et al., 2000)
1.2 Nicotinic Acetilcholine Receptors (nAChRs)
Neuronal nAChR is ligand-gated cation channels that was activated by the endogenous neurotransmitter acetylcholine (ACh) and the exogenous tertiary alkaloid nicotine (Albuquerque et al., 2009). nAChRs are the superfamily of Cys-loop ligand gated ion channels that include receptors for γ-amino butyric acid (GABA, the GABAA, and
GABAC receptor), glycine, and 5-hydroxytryptamine (5-HT3), invertebrate glutamate and
histidine receptors (Tsunoyama and Gojobori, 1998). Throughout of the subunits in this family contain a pair of disulfide-bonded cysteines separated by 13 residues (Cys-loop) in their extracellular amino terminus (Karlin, 2002).
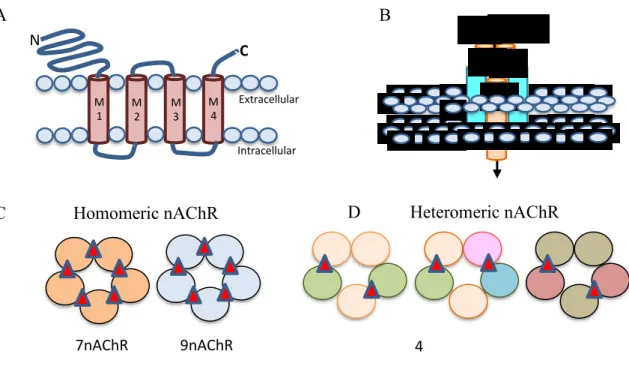
The nAChR gene encodes a protein subunit consisting of a large amino-terminal extracellular domain composed of β-strands, four transmembrane α-helices segments (M1-M4), a variable intracellular loop between M3 and M4 as a prominent variable loop, and an extracellular carboxy-terminus in figure 2A (Corringer, Novère and Changeux, 2000). The extracellular N-terminus contains the ACh binding domain that forms a hydrophobic pocket located between adjacent subunits in an assembled receptor (Sine, 2002). The M2 segment of all five subunits forms, coassemble to form a functional subunit, the conducting pore of the channel, and regions in the M2 intracellular loop contribute to cation selectivity and channel conductivity (Corringer, Novère and Changeux, 2000) figure 2B. Subunits are classified as either α, by the presence of a Cys-Cys pair near the start of TM1, or non-α (β)
6
when the Cys pair is missing (Changeux and Edelstein, 1998) Five subunits combine to form two classes of receptors: homomeric receptors containing only α subunits (α7-α9), homomeric receptor usually have low affinity for agonist. Heteromeric receptors that contain α and β subunits (α2-α6 and β2-β4) (Dani and Bertrand, 2006) figures 2C,D. The most abundant subtypes in the brain are the low affinity α7 homomeric and high affinity α4β2∗ heteromeric nAChRs. ACh binding sites are depicted as red triangles.
Figure 2. Neuronal nAChR Structure.(Hendrickson, Guildford and Tapper, 2013)
The nAChRs receptors are critical for drug addiction through stimulation of synaptic activity in the hippocampus, amygdala, ventral tegmental area (VTA) and nucleus accumbens (NAc) (De Biasi and Dani, 2011; Leslie, Mojica and Reynaga, 2013). The α7 homo-oligomer and α4β2 hetero-oligomer are two major subtypes of nAChRs in the mammalian brain (Changeux, 2010b) Both of that receptor regulates the nicotine dependence, chronic nicotine exposure selectively up-regulates the density of α4β2 to elevates nicotine addiction in rats (Nguyen, Rasmussen and Perry, 2003). Synergy with
M 1 M 2 M 3 M 4 C N Extracellular Intracellular α4 α4 β2 β2 β2 Na+ Ca2+ A B β2 β2 β2 β2 β2 β3 β4 β4 β4 α4 α4 α4 α6 α3 α3 α 7 α 7 α 7 α 7 α 7 α 9 α 9 α 9 α 9 α 9
C Homomeric nAChR D Heteromeric nAChR
7
α4β2 the α7 homo-oligomer also involved in nicotine dependence, evidenced by the high expression of α7nAChR on VTA in a nicotine dependence rat model (Nomikos et al., 2000).
8
CHAPTER 2
INHIBITION OF NICOTINE DEPENDENCE BY CURCUMINOID IS ASSOCIATED WITH REDUCED ACETYLCHOLINESTERASE ACTIVITY IN
THE MOUSE BRAIN 2.1 Introduction
2.1.1 Acetylcholine
Acetylcholine (ACh) is a fast acting and point to point neurotransmitter at the neuromuscular junction and in the autonomic ganglia, there are fewer demonstrations of similar actions in the brain (Changeux, 2010a)
.
ACh critical for the response to uncertainty, such that an increase in cholinergic tone predicts the unreliability of predictive cues in a known context and improves the signal to noise ratio in a learning environment (Yu and Dayan, 2005).
Acetylcholine in the brain alters neuronal excitability, influences synaptic transmission, induces synaptic plasticity, and coordinates firing of groups of neurons. As a result, it changes the state of neuronal networks throughout the brain and modifies their response to internal and external inputs actions of cholinergic signaling on cellular and synaptic properties of neurons in several brain areas and discuss consequences of this signaling on behaviors related to drug abuse, attention, food intake (Picciotto, Higley and Mineur, 2012).
ACh signals through two classes of receptors: metabotropic muscarinic receptors (mAChRs) and ionotropic nicotinic receptors (nAChRs) (reviewed in Picciotto et al., 2000 and Wess, 2003a). Muscarinic receptors are coupled either to Gq proteins (M1,
M3, and M5 subtypes) that activate phospholipase C or Gi/o proteins (M2 and M4 subtypes)
9
a variety of biochemical signaling cascades. Moreover, mAChRs are located both pre- and postsynaptically throughout the brain, producing diverse consequences for brain activity (Figure 3).
Figure 3. Sites of Action for Acetylcholine, Nicotinic and Muscarinic Acetylcholine Receptors (Picciotto, Higley and Mineur, 2012)
Acetylcholine were hydrolyzed and inactivated by Acetylcholinesterase (AChE ), thereby regulating the concentration of the transmitter at the synapse. Termination of activation is normally dependent on dissociation of acetylcholine from the receptor and its subsequent diffusion and hydrolysis, except in diseases where acetylcholine levels are limiting or under AChE inhibition, conditions that increase the duration of receptor activation(SILVER, 1963).
10
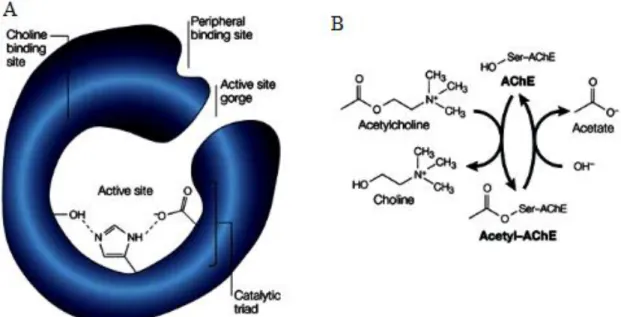
Figure 4. a. Structural feature of the AChE enzyme. X-ray crystallography has
identified an active site at the bottom of a narrow gorge, lined with hydrophobic amino -acid side chains. At the time, the catalytic triad was unique among serine hydrolases in having a glutamate side chain in lieu of the familiar aspartate side chain. A choline-binding site featured hydrophobic tryptophan residues instead of the expected anionic groups; a peripheral binding site has also been identified by site-directed mutagenesis. b.
The acetylcholinesterase (AChE) reaction. AChE promotes acetylcholine hydrolysis
by forming an acetyl-AChE intermediate with the release of choline, and the subsequent hydrolysis of the intermediate to release acetate (Silver, 1963).
Since AChE is key enzyme responsible for metabolizing acetylcholine in synapse (Subramaniyan and Dani, 2015), inhibition of AChE activity increase augment cholinergic transmission and extracellular acetylcholine, recents study showed galantamine an acetylcholinesterase inhibitor attenuates nicotine taking and seeking in the rat (Hopkins et
al., 2012) and restore cognitive function on nicotine withdrawal in mice C57BL/6
11
nicotine seeking and reward in the rat due to drug-induced motor suppressant effects or increased adverse effects (Kimmey et al., 2014)
2.1.2 Curcuminoids
Curcuminoid is a compound in turmeric that is derived from the rhizome and grounded roots of Curcuma longa. Curcuminoids are composed of curcumin (77%), desmethoxycurcumin (17%), and bisdemethoxycurcumin (3%)(Prasad et al., 2014), the chemical structure were showed on Figure 3. Curcumin reduces inflammation via suppression of IL1-induced NF-B activation in the cultured human tendon (Buhrmann et
al., 2011). Furthermore, curcumin treatment inhibits H2O2-induced phosphorylation of p38
and JNK, which are pro-apoptotic signals, and restores the phosphorylation of ERK and Akt in PC12 cells. This suggests that curcumin has anti-inflammatory effects that can prevent apoptosis (Fu et al., 2016). In addition, Dzoyem and Elof (2015) report that anti-inflammatory compounds, such as a phenolic compound (curcuminoid) or flavonoid, inhibit acetylcholinesterase (AChE) activity in vitro (Dzoyem and Eloff, 2015).
Figure 5. Curcuminoids Consists of Curcumin, Bisdemethoxycurcumine and Desmethoxycurcumin (Paramasivam et al., 2009)
O O HO OH O O Curcumin HO OH O O Bisdemethoxycurcumin O HO OH O O Desmethoxycurcumin
12
2.2 Objectives
The aim of the current study was to assess the ameliorating effect of curcuminoid on nicotine dependence and its effect on AChE activity. Furthermore, we compare the effect of curcuminoid by the AChE inhibitors, donepezil.
13
2.3 Materials and Methods 2.3.1 Animals and Materials
Male Swiss-Webster mice aged 8 weeks (20–30 g) were used in all experiments (School of Pharmacy, Institute Technology of Bandung). Mice were housed five per cage in a room with a 12/12 hour light/dark cycle (lights on at 08:00). Room conditions were temperature controlled at 22.0 ± 2 °C with a relative humidity of 50% ± 20%. Mice had free access to food and water prior to the start of the experiment. All experiments were approved by the Ethics for Animal Care and Use board.
2.3.2 Drug administration
Nicotine (0.5 mg·kg-1, Sigma Aldrich) was dissolved in saline 0.9%, donepezil (1,
3.2, and 10 mg·kg-1, wako), was dissolved in distilled water, and curcuminoid was
suspended in 1% NaCMC. Mice were divided into four groups: control (n = 5), nicotine (n = 5), nicotine + curcuminoid (n = 5), nicotine + donepezil (n=5). All mice were habituated to the conditioned placed preference (CPP) box for 5 days. Following this, mice were pre-conditioning, where the nicotine compartment was paired with the least preferred side of the compartment (Carboni and Vacca, 2002). Mice received treatment 7 days after the pre-conditioning. Briefly, control mice received a saline injection (i.p.) and were confined to the designated compartment for up to 30 minutes per day. Mice in the nicotine group received a nicotine injection (0.5 mg·kg-1, i.p.) followed by placement in the nicotine paired
compartment for 30 minutes. Mice in nicotine + curcuminoid, nicotine + donepezil groups received an oral dose (1, 3.2, or 10 mg·kg-1) of curcuminoid or donepezil 30 minutes prior
to nicotine administration followed by confinement in the designated CPP compartment for 30 minutes. Four hours after treatment, all mice were received a saline injection and placed in the opposite CPP compartment.
14
The CPP test was performed 1 day after the conditioning phase. The place preference score of each mouse was recorded determine dependence. Mice underwent an extinction phase for 5 days after the CPP test, where no nicotine was administered. The next day, the CPP test was repeated twice to assess the preference of the mice after extinction. First, place preference was recorded without any drug administration to confirm extinction. Next, 0.5 mg·kg-1 nicotine was administered to nicotine, nicotine + curcuminoid groups to induce
relapse. Mice nicotine + curcuminoid, nicotine + donepezil. Donepezil orally prior to nicotine administration.
2.3.3 Conditioned Place Preference Test
The conditioned place preference test was performed using a biased design with four phases: habituation (5 days), conditioning (7 days), extinction (5 days), and relapse.
Figure 6. Scheme of the experiment, mice were habituated to the CPP apparatus for 5
days, followed by a pre-conditioning test to determine which compartment would be paired with the drug administration. Following this, the mice were conditioned for 7 days. Each day they were administered with saline (control), 0.5 mg·kg-1 nicotine (nicotine), 0.5 mg·kg
-1 nicotine and 1 mg·kg-1, 3.2 mg·kg-1, and 10 mg·kg-1 curcuminoid (nicotine + curcuminoid),
0.5 mg·kg-1 nicotine and donepezil 1 mg·kg-1, 3.2 mg·kg-1, and 10 mg·kg-1 (nicotine +
donepezil), followed by confinement in the designated compartment. One day after conditioning, the preference ratio was measured to assess nicotine dependence. Mice
day 18 Day 0
Acclimatization Conditioning Extinction
Preconditioning test Day 5 Conditioning test Day 12 Abstinence test Day 17 Nicotine ± Curcuminoid or Donepezil (1, 3.2, 10 mg/kg) Saline Priming test Day 18 Dissection Nicotine ± Curcuminoid or Donepezil (1, 3.2, 10 mg/kg)
15
entered the extinction phase, when no drugs were administered, for 5 days. The preference ratio was recorded after 5 days to confirm the extinction. A single dose of nicotine was administered the next day to measure relapse. Mice brains were extracted for measurement of acetylcholinesterase activity
2.3.4 Habituation
Acclimatization was performed for 5 days prior to pre-conditioning. It is designed to remove any environmental stress, including the weighing and testing rooms, CPP apparatus, and drug administration.
2.3.5 Pre-conditioning test
The pre-conditioning test was used to determine which compartment should be drug or nicotine-paired. The compartment with the lower ratio preference was used [15]. One day after habituation, each mouse was placed in the grey compartment for 5 minutes with closed guillotine door. Following this, the guillotine door was opened and the mouse could access all compartments for 15 minutes. Time spent in each compartment was recorded and preference ratio was calculated using following formula:
𝑃𝑟𝑒𝑓𝑒𝑟𝑒𝑛𝑐𝑒 𝑟𝑎𝑡𝑖𝑜 = 𝑆𝑜𝑗𝑜𝑢𝑟𝑛 𝑡𝑖𝑚𝑒 𝑖𝑛 𝑟𝑒𝑙𝑎𝑡𝑒𝑑 𝑐𝑜𝑚𝑝𝑎𝑟𝑡𝑚𝑒𝑛𝑡 (𝑠)
𝑇𝑜𝑡𝑎𝑙 𝑡𝑖𝑚𝑒 𝑠𝑝𝑒𝑛𝑡 𝑖𝑛 𝑎𝑙𝑙 𝑐𝑜𝑚𝑝𝑎𝑟𝑡𝑚𝑒𝑛𝑡𝑠 (𝑠) × 100
2.3.6 Conditioning and Post-conditioning testing
Mice were conditioned for 7 days. Each day they were injected with nicotine (nicotine, nicotine + curcuminoid, nicotine + donepezil groups) or saline (control group) followed by confinement in the designated compartment (Benowitz et al., 1988a). Nicotine + curcuminoid and nicotine + donepezil groups received an oral dose (1, 3.2, or 10 mg·kg
-1) of curcuminoid or donepezil 30 minutes prior to nicotine administration. Four hours later,
the same procedure was repeated with saline administration for all groups. Mice were then confined to the opposite compartment of the apparatus. The next day, the conditioning
16
preference ratio was calculated to assess nicotine dependence. One day later, the conditioning preference ratio for each mouse was measured to confirm extinction and relapse using following formula:
𝑃𝑟𝑒𝑓𝑒𝑟𝑒𝑛𝑐𝑒 𝑟𝑎𝑡𝑖𝑜 = 𝑆𝑜𝑗𝑜𝑢𝑟𝑛 𝑡𝑖𝑚𝑒 𝑖𝑛 𝑟𝑒𝑙𝑎𝑡𝑒𝑑 𝑐𝑜𝑚𝑝𝑎𝑟𝑡𝑚𝑒𝑛𝑡 (𝑠)
𝑇𝑜𝑡𝑎𝑙 𝑡𝑖𝑚𝑒 𝑠𝑝𝑒𝑛𝑡 𝑖𝑛 𝑎𝑙𝑙 𝑐𝑜𝑚𝑝𝑎𝑟𝑡𝑚𝑒𝑛𝑡𝑠 (𝑠) × 100
2.3.7 Extinction and abstinence test
The extinction and abstinence test was carried out 1 day after the post-conditioning tests. Mice were injected with saline for 5 days during the extinction phase; therefore, both conditioning compartments were paired with saline (Natarajan, Harding and Wright, 2013). One day later, the preference ratio for abstinence was calculated using the same procedure as the pre- and post-conditioning tests.
2.3.8 Priming nicotine/relapse
Mice were injected with 0.5 mg·kg-1 nicotine and the place preference ratio was
calculated using the same procedure as the pre- and post-conditioning tests.
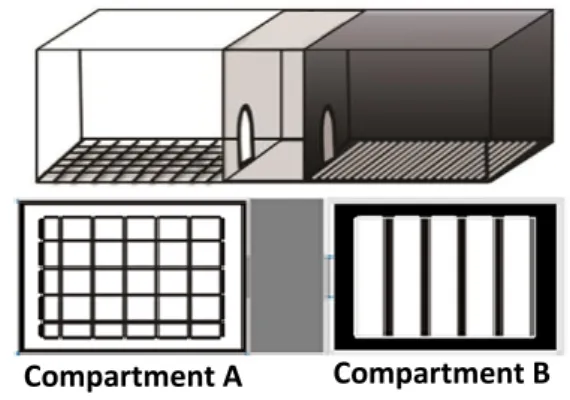
2.3.9 CPP (Conditioned Place Preference) Apparatus
Figure 7. Conditioned Place Preference Box
The apparatus for the conditioned place preference test consisted of three compartments measuring 12.7 cmx46.5 cmx12.7 cm (width x length x height) in size. The middle compartment was grey, called neutral compartment. Two conditioning
17
compartments differed in color and floor texture. Compartment A was white with a quadrangular sieve (mesh). The other compartment (B) was black with stainless steel floors. Each compartment was separated by two doors.
2.3.10 Acetylcholinesterase activity assay
Ellman's method was used to determine AChE activity. Brain samples of mice were isolated immediately after the priming tests.
2.3.10.1 Brain sample preparation
Mice were sacrificed by decapitation and the brain was isolated, weighed, and washed with saline. Brains were stored at –70°C for further analysis (Trudeau and Cartier, 2000). Tissue was homogenized in 0.05 M phosphate buffer (pH 7.2) using an Edmund Bühler homogenizer and equalized to a total protein concentration of 20 mg·ml-1. Following
this, aliquots were incubated at 37 °C for 10 minutes. Any insoluble material was removed by centrifugation (10000 g) for 5 minutes. Each sample (400 μl) was mixed with 2.6 ml phosphate buffer, 10 μl acetylcholine chloride, and 20 μl dithiobis-nitrobenzoic acids (DTNB) to assess AChE activity (Ellman et al., 1961).
2.3.10.2 Assay of enzyme activity
Enzyme activity was measured using a spectrophotometer (Beckman Coulter DU-720) at a wavelength of 412 nm. We used a kinetic model in which the absorbance was measured for 6 minutes at 1-minute intervals:
Enzyme activity was calculated using the following formula (Ellman et al., 1961): R = 5.74 × 10-4 × ΔA/Co where,
R: the rate of substrate hydrolysis (mol·min·g-2 brain tissue) ΔA: Change in absorbance per minute
18
2.3.11 Statistical analysis
Data were analyzed using Statistical Package for the Social Sciences (SPSS) software version 18. After analysis of variance (ANOVA) tests were conducted, post-hoc Tukey LSD was used to control for multiple comparisons (Kim, 2015). Differences were considered significant when p <0.05.
19
2.4 Results
2.4.1 Nicotine administration generates nicotine dependence and relapse in mice
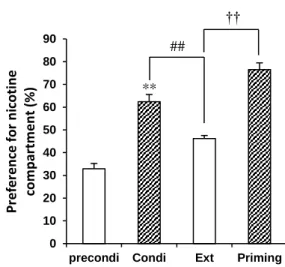
In pre-conditioning phase, a single nicotine injection induced a preference ratio of 31.9% . Figure 8 shows that 7 days of nicotine injections creates nicotine dependence in a mouse. Repeated nicotine administration (0.5 mg·kg-1) significantly increased the
preference ratio to 63.5% (p < 0.01). By contrast, saline injections (during the extinction phase) significantly decreased the preference ratio to 46.2% (p < 0.01) when compared with the conditioning phase. Interestingly, nicotine administration following extinction significantly enhanced the preference ratio to 76.515% (p < 0.01) when compared with the extinction phase.
Figure 8. Nicotine induced dependence. Preconditioning = pretreatment phase without
nicotine administration (5 days), Condi = conditioning phase where mice received nicotine 0.5 mg·kg-1 for 7 consecutive days. Preference ratio was recorded on day 8. Ext = extinction
phase, no nicotine was administered (5 days). Preference ratio was recorded the following day. Priming = mice were challenged with an administration of 0.5 mg·kg-1 nicotine to
induce relapse. Data = mean ± SEM, n = 10. **p < 0.01 when compared with
pre-** 0 10 20 30 40 50 60 70 80 90
precondi Condi Ext Priming
Pr e fer e n ce for n ic o tine co m p ar tm e n t (% ) ## ††
20
conditioning treatment. ##p < 0.01 when compared with conditioning treatment. ††p < 0.01
when compared with abstinence treatment.
2.4.2 Curcuminoid attenuates the nicotine-induced preference ratio in the CPP test at conditioning and priming phases
Importantly, pre-administration with 1, 3.2, or 10 mg·kg-1 curcuminoid 30 min prior
to nicotine administration reduced the preference ratio in a dose-dependent manner (preference ratio, 1 mg·kg-1 curcuminoid: 46.9%, p < 0.05; 3.2 mg·kg-1 curcuminoid: 45.9%,
p<0.05; 10 mg·kg-1 curcuminoid: 41.32%, p < 0.01; when compared with nicotine group).
The effect of curcuminoid comparable with the donepezil 1 mg·kg-1 42.62% p>0.05,
donepezil 3.2 mg·kg-1 38.93% p>0.05, donepezil 10 mg·kg-1 10 mg/kg 38.6% p>0.05 on
prevents nicotine dependence Taken together, these results show that 0.5 mg·kg-1 nicotine
induces nicotine dependence and relapse in mice, which is ameliorated by administration of curcuminoid. ** # # ## ## ## ## 0 10 20 30 40 50 60 70 80 Pr e fer e n ce for n ic o tine co m p ar tm e n t (% ) Curcuminoid Donepezil
7 days nic conditioning (test at day 12 of experiment)
- + + + + - - 1 3.2 10 Nicotine 0.5 mg/kg Curcuminoid or Donepezil (mg/kg) NS NS NS
21
Figure 9. Preference ratio following 7 days of conditioning. After in all four treatments
group the x-axis shows the administration protocol for each group. One group was treated without nicotine (control). Data represent the median ± SEM. n = 5 in each group. **p < 0.01 when compared with control group. ##p < 0.01, #p < 0.05 when compared with the
nicotine group at the conditioning phase. NS = no significant different curcuminoid vs donepezil
2.4.3 Nicotine-induced Withdrawal is attenuated by pre-administration with curcuminoid
Figure 10. The preference for nicotine following 5 days of abstinence. Data are
expressed as the median ± SEM. n = 5 in all groups. **p < 0.01 when compared with the control group. ##p < 0.01 when compared with the nicotine group at the abstinence phase.
** ## ## ## ## ## 0 10 20 30 40 50 60 Pr e fer e n ce for n ic o tine c o m p ar tm e n t (% ) Curcuminoid Donepezil
Extinction test at day 17
- + + + + - - 1 3.2 10 Nicotine 0.5 mg/kg Curcuminoid or Donepezil (mg/kg) NS NS NS
22
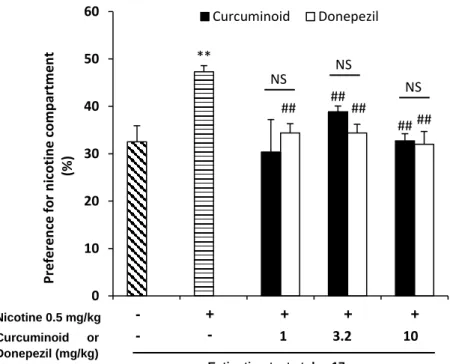
We next evaluated the effect of curcuminoid on extinction (Figure 10). All groups underwent an extinction phase (abstinence) for 5 days after conditioning. Mice were injected with saline only. The preference ratio after the extinction phase in the nicotine + curcuminoid group was significantly reduced when compared with nicotine only group [preference ratio: nicotine = 47.3%; vs nicotine + curcuminoid (3.2 mg·kg-1) = 38.%, p <
0.01; vs nicotine + curcuminoid (10 mg·kg-1) = 32.7%, p < 0.01]. Donepezil also reveals the
identical phenomena with the curcuminoid group on extinction phase p>0.05. preference ratio donepezil (1 mg·kg-1 34% p<0.01), (3.2 mg·kg-1 34.39% p<0.01), (10 mg·kg-1 32.01%
p<0.01) significantly difference with the nicotine group treatment (47.3%).
2.4.4 Nicotine-induced relapse is attenuated by pre-administration with curcuminoid
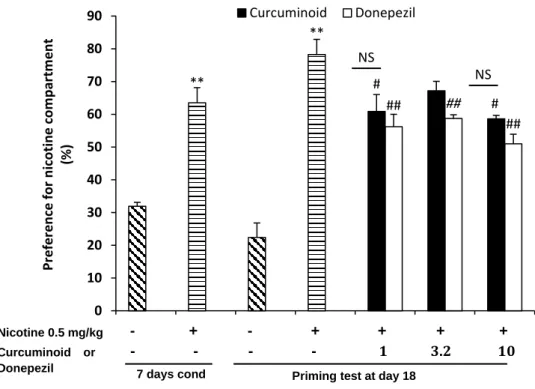
Furthermore, we assessed the effect of curcuminoid on relapse behaviors. Figure 11
shows the curcuminoid administration successfully attenuated relapse as well as donepezil administration, as measured by a significant reduction in preference ratio [preference ratio: nicotine = 78.25%; vs 1 mg·kg-1 curcuminoid (60.9%), p < 0.05; vs 10 mg·kg-1 curcuminoid
(58.6%), p < 0.05; Nicotine vs 1 mg·kg-1 donepezil (56.22%), p<0.01; 3.2 mg·kg-1 donepezil
23
Figure 11. Preference for nicotine following relapse when 0.5 mg·kg-1 nicotine was
administered to all mice. Curcuminoid was administered 30 minutes prior to nicotine
administration. The preference ratio was recorded after nicotine administration. Data are expressed as mean ± SEM. n = 5 in all groups. **p < 0.01 when compared with the control group in each phase. #p < 0.05 when compared with the nicotine group, ## p < 0.01 when
compared with the nicotine group, and †p < 0.05 curcuminoid vs donepezil in the priming
phase. NS = no significant different curcuminoid vs donepezil.
2.4.5 Curcuminoid inhibits acetylcholinesterase activity during nicotine reward behavior
Figure 12 shows the effect of curcuminoid on nicotine-induced AChE activity. Nicotine administration significantly upregulated AChE activity when compared with the control group (p < 0.01). Curcuminoid significantly attenuated the nicotine-induced AChE
** ** # # ## † ## ## 0 10 20 30 40 50 60 70 80 90 Pr e fer e n ce for n ic o tine c o m p ar tm e n t (% ) Curcuminoid Donepezil
Priming test at day 18
- + - + + + + Nicotine 0.5 mg/kg 7 days cond Curcuminoid or Donepezil NS NS - - - - 1 3.2 10
24
activity. Moreover, there was no difference in AChE activity between 3.2 mg·kg-1 and 10
mg·kg-1 curcuminoid and control groups (3.2 mg·kg-1 curcuminoid vs control, p = 0.0510;
10 mg·kg-1 curcuminoid, p= 0.0939).
Figure 12. Curcuminoid inhibits acetylcholinesterase activity in the mouse brain
Ellman’s method was used to measure acetylcholinesterase activity in the mice brains. Data are expressed as mean ± SEM. n = 4–6 mice in each group. **p < 0.01, *p < 0.05 when compared with the control group. ##p < 0.01, #p < 0.05 when compared with the nicotine
treatment groups. Nicotine 0.5 mg/kg - + + + + ** * # ## ## 0.28 0.29 0.3 0.31 0.32 0.33 0.34 A ctiv ity o f A Ch E Curcuminoid (mg/kg) - - 1 3.2 10
25
2.5 Discussion
This study shows that 7 days of nicotine administration is sufficient to induce dependence in mice, but this can be attenuated by curcuminoid or donepezil. Moreover, it prevents nicotine relapse following 5 days of abstinence. We hypothesize that these compounds prevent dependence and relapse via inhibition of AChE activity, as curcuminoid reduce and fully attenuate, respectively, the nicotine-induced increase in AChE activity.
CPP is most popular method to evaluate reward effect of nicotine (Carboni and Vacca, 2002). This method is substantially different from the drug self-administration method (Bardo and Bevins, 2000). It is a beneficial method to use when seeking a monophasic dose-response curve, whereas self-administration experiments elicit inverted U-shaped dose-response curves. In the drug administration method, repeated self-infusions are required to establish reliable behavior. It is likely that repeated exposure affects receptor transduction mechanisms associated with tolerance and sensitization (Bardo and Bevins, 2000). Moreover, CPP is the preferred method for rapid screening and can be used with many mouse strains with high sensitivity (Cunningham et al., 1999).
Prolonged nicotine exposure results in neural adaptation following receptor desensitization and an upregulation of nAChRs (Benowitz, 2008). Nicotine enhances dopamine (DA) release in striatal cholinergic interneurons in mice, as measured by fast-scan cyclic voltammetry, and this is correlated with nicotine dependence (Mclaughlin, Dani and Biasi, 2015). In addition, nicotine promotes ROS generation in a dose-dependent manner in rat mesencephalon cells; nitric oxide (NO) and peroxide (H2O2) damage cell
membranes that induces tissue damage. Curcuminoid is an antioxidant; curcuminoid administration reduces the production of superoxides by macrophages, which are activated with phorbol-12-myristate-13-acetate (PMA), in mice (Ruby et al., 1995). Moreover,
26
demethylated curcuminoid and curcumin, derived from curcuminoid, inhibit the glutamate-induced generation of cellular ROS without elevating intracellular calcium in mouse hippocampal neuronal cells (Khanna et al., 2009). ROS production in rat NAc neurons has been shown to contribute to drug addiction. These data suggest that curcuminoid may be inhibiting of nicotine dependence via inhibition of nicotine-induced ROS generation (Jang
et al., 2015).
Additionally, through ROS-induced oxidative stress enhanced the activity of AChE, compromise of the enzyme activity have an important role in the maintenance of acetylcholine synaptic levels, thus preventing or improving cognitive and memory functions (Melo, Agostinho and Oliveira, 2003). Recent reports showed, that an increase in cell Ca2+ influx, mainly through voltage-sensitive Ca2+ channels, precedes ROS formation and
the subsequent lipid peroxidation in cultured cells and stimulates AchE activity (Ekinci, Linsley and Shea, 2000).
It has been hypothesized that curcuminoid can attenuate nicotine dependence via inflammatory mechanisms. A previous study found a correlation between the reward and inflammatory systems via arachidonic acid (ARA): tetrahydrocannabinol increases prostaglandin, an ARA precursor, which attenuated addiction from diclofenac sodium (10 mg·kg-1, i.p.) in mice (Anggadiredja et al., 2004). Non-steroidal anti-inflammatory drugs
(NSAIDs) inhibit fatty acid amide hydrolase (FAAH) or other possible intracellular transporters of endocannabinoids, which prevents active constituent binding by CB1 cannabinoid receptors in many brain areas. This reduces the reward of cannabinoids (Păunescu H et al., 2011). Moreover, pharmacological blockade of FAAH in the rat significantly inhibits nicotine reward, but not nicotine withdrawal (Muldoon et al., 2013).
27
Nicotine increases brain acetylcholinesterase activity in a context-dependent manner in zebrafish (Ziani et al., 2018). Supposedly, the upregulation of AChE activity by nicotine was started by stimulating Ach release since the hippocampal formation receives its dopaminergic inputs from the VTA (Scatton et al., 1980; Umegaki et al., 2001) the nicotine-induced hippocampal ACh release may be mediated, in part, via the activation of nAChRs located in the VTA. Dopaminergic regulation of hippocampal ACh release is established from anatomical and functional studies as summarized below.
Hippocampal dopamine receptors have been localized to the molecular layer of the dentate gyrus and the dorsal hippocampus (Tiberi et al., 1991). Significant loss of hippocampal dopamine receptors with a concomitant loss of choline acetyltransferase activity (ChAT) after fimbriaectomy (Hersi et al., 1995) suggests that a subpopulation of the dopamine receptors are presynaptically localized on the cholinergic afferents. These observations, taken together, further support the involvement of nAChR activation of VTA-hippocampal DA-ergic pathway, DA release, dopamine receptor activation in nicotine-induced hippocampal ACh release in vivo. The abundance of Ach in brains leads to increase acetylcholinesterase activity to degrade the Ach to choline and acetate (Dvir et al., 2010).
It has also been hypothesized that curcuminoid attenuates nicotine dependence via inhibition of AChE activity pathway. Recent data have shown galantamine and donepezil inhibit of AChE, which prevents nicotine seeking and reward in the rat. This could be due to the drug-induced suppression of motor functions or an increase in adverse effects (Hopkins et al., 2012; Kimmey et al., 2014).The mechanism of curcuminoid to reduce the main effect of nicotine through AChE inhibition may partially understand by evidence indicating AChEIs substitute for the discrimination stimulus of nicotine (Giarola, Auber and Chiamulera, 2011). Curcuminoid alleviates nicotine taking by producing a subjective effect
28
similar to nicotine (Giarola, Auber and Chiamulera, 2011). The interoceptive properties of nicotine are mediated primarily by α4β2 nAChRs (Shoaib, Zubaran and Stolerman, 2000; Zaniewska et al., 2006). AChEIs as positive allosteric modulators of α4β2 nAChRs (Maelicke and Albuquerque, 2000), when combined with a sub-threshold dose of nicotine, produce nicotine-like discriminative stimulus effects (Zaniewska et al., 2006) to alleviates nicotine dependence.Thus, it is possible that AChE inhibition by curcuminoid may increase or enhance the interoceptive cues of nicotine by activating α4β2 nAChRs thereby reducing subsequent nicotine consumption. Additionally, proposed that the effect of acute AChE inhibition on nicotine self-administration in rats indicates that increased cholinergic transmission is a prerequisite for the elimination of nicotine reinforcement (Ashare et al., 2016), and repairing cognitive impairment (Marcos et al., 2009).
Donepezil was used a reference substance in this experiment, Donepezil (C24H29NO3,
MW: 379.492) is a reversible, selective AChE inhibitor that is currently approved for the symptomatic treatment of Alzheimer disease (Castro and Martinez, 2006; Arce et al., 2009; Colovic et al., 2013). Previous research reveals Donepezil, an acetylcholinesterase inhibitor, attenuates nicotine self-administration and reinstatement of nicotine seeking in rats[14]. Cholinergic transmission increased by donepezil in the brain through inhibition of acetylcholinesterase, thereby increasing endogenous acetylcholine levels (Giacobini et al., 1996; Kosasa, Kuriya and Yamanishi, 1999) [42,43]. Previous research has shown that AChE inhibitor administration promotes discriminative stimulus properties of nicotine, which suggests that these compounds share similar interceptive stimulus characteristic [33]. Therefore, donepezil reduces nicotine taking, through increasing endogenous acetylcholine levels in the brain to produce subjective effects similar to nicotine. Another base evidence for supporting this hypothesis is demonstrating that the α4β2 nicotinic acetylcholine
29
receptor partial agonist varenicline is generalizing to the discriminative stimulus properties of nicotine (Rollema et al., 2007)
Previous studies have reported that curcumin decreases AChE activity in rat brain (Peeyush Kumar et al., 2011; Khurana et al., 2012; Tüzmen et al., 2015). In vitro experiments also confirmed that curcumin inhibits AChE activity and has an IC50 value of
67 µM. The strongest AChE inhibition was found using bisdemethoxycurcumin; a single dose of 30 µM of bismethoxy curcumin, dimethoxy curcumin, or curcumin inhibits 85%, 55% or 31% of AChE activity, respectively (Ahmed and Gilani, 2009). Additionally, molecular docking study (in silico) shows curcumin a component of curcuminoid interacts with actives site of AChE on amino acids (Tyr133, Glu202, Phe295, and Tyr337) to disrupt the activity of AChE (Saravanan, Kalaiarasi and Kumaradhas, 2017).
Dopaminergic pathway were proposed involved on curcuminoid prevents nicotine dependence, curcumin reduced upregulation of dopamine D1 and dopamine D2 receptor induced by STZ in diabetes mice models, through CREB and Phospholipase C expression regulation in cerebral cortex and cerebellum and thereby improving the cognitive and emotional functions associated with these regions (Kumar et al., 2010). The pivotal role of D1R on nicotine dependence were confirmed by administration of D1R antagonist SCH-23390 in NAc, ACC and PtA inhibits nicotine-induced behavior including dependence in mice (Kutlu et al., 2013; Hall et al., 2015a), and eticlopride injection an antagonist of D2R reduced the ability of nicotine to enhance the responding for conditioned reinforcement effect (Guy and Fletcher, 2014). Unfortunately, donepezil as a drug reference not affected on dopaminergic neuron were proven by unchangeable the dopamine striatal regulation after donepezil administration (Reeves et al., 2010).
30
In summary, we have shown that curcuminoid blocks the development of nicotine dependence and relapse and this is mediated in part by the enhancement of the cholinergic system. Most studies have focused on nicotine reward circuits in nicotine dependence in animal models. These circuits include many brain regions, such as the ventral tegmental area (VTA), nucleus accumbens, and prefrontal cortex, which modulate dopamine signaling to promote dependence (Wise, 2002). The present study has revealed a new association with nicotine reward circuits through the cholinergic system. However, further studies are required to further elucidate the anti-inflammatory role and curcuminoid-induced dopaminergic signal in the amelioration of nicotine dependence.
31
CHAPTER 3
CRUCIAL ROLE OF DOPAMINE D2 RECEPTOR SIGNALING IN NICOTINE-INDUCED CONDITIONED PLACE PREFERENCE
3.1 Introduction
3.1.1 Nicotine Reward System
Reward is experienced in response to discrete stimuli, providing enjoyment and arousal, furthermore addictions involve persistent, compulsive, and uncontrolled behaviors that are both maladaptive and destructive (Adinoff, 2004), reward and addiction obviously not same but thought to share common neurobiology underpinning.
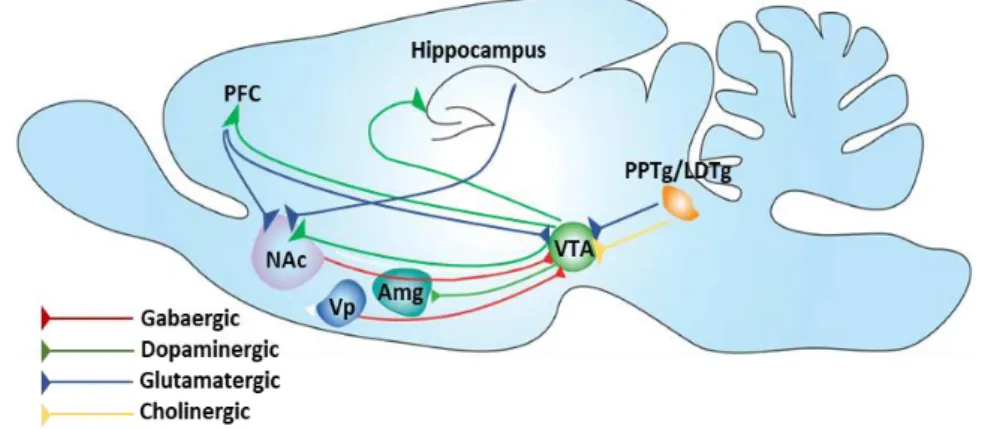
Figure 13. Nicotine reward system (Subramaniyan and Dani, 2015).
Firstly VTA receives convergent inputs from many areas, but the prominent input sources contributed to reward-based learning mostly from pedunculopontine/lateral dorsal tegmentum (PPTg/LDTg) (Oakman et al., 2018) and the PFC (Carr and Sesack, 2000). The PPTg sends glutamatergic/cholinergic projections to the VTA, and activation of PPTg elicits burst firing in VTA DA neurons (Floresco et al., 2003). VTA consists of heterogeneous group of cells: the paranigral nucleus, the parainter fascicular nucleus, the parabrachial pigmented nucleus, and the rostral VTA, it is located bilaterally near the midline at the floor of the midbrain (Fig. 13) (Oades and Halliday, 1987). It is an important node in the
reward-32
based learning circuit (Fig. 13,) and projects to many brain structures including the NAc, PFC, and hippocampus (Swanson, 1982).
The VTA dopaminergic neurons spontaneously fire at a tonic average rate of around 4 Hz (2–10 Hz range) with randomly interspersed bursts (Grace and Bunney, 1983). During the acquisition phase of reward-based learning, they fire briefly in phasic bursts, signaling and broadcasting the arrival of reward (Ljungberg, Apicella and Schultz, 1992). Once the association between reward predicting cues and rewards is learned, the arrival of reward no longer elicits burst firing. Instead, the conditioned incentive stimuli that predict reward elicit burst firing (Montague, Dayan and Sejnowski, 1996). When nicotine arrives in the brain, among its many actions is to excite the midbrain dopaminergic centers of the VTA and SNc (Zhang et al., 2009). Nicotinic receptors are found on the GABAergic interneurons and DA neurons within the VTA and on the excitatory and inhibitory input axons and/or terminals. Activation of nAChRs located on VTA DA neurons be essential for nicotine addiction in animal models (Picciotto et al., 1998).
3.1.2 Nucleus Accumbens
Figure 14. Nucleus Accumbens (Subramaniyan and Dani, 2015)
One of the key targets that receives potent nicotine- induced DA release from the VTA is the NAc. The NAc is divided into core and shell sub-regions based on anatomy and
33
neurochemical make up. The core region is surrounded on the medial, lateral, and ventral sides by the shell region (Fig. 14). On the dorsal side, the core is directly continuous with the dorsal part of the striatum. In many respects, the core boundary cannot be easily distinguished from the dorsal part of the striatum. The core and shell regions receive distinct but overlapping inputs. Anatomical connections also exist between the core and shell, allowing interactions between them. Given the distinction between the core and the shell, many studies have explored the relative role of these two regions in instrumental learning in general and nicotine addiction in particular
Nicotine modulates DA levels in the NAc in two ways. First, nicotine activates VTA neurons, which leads to DA release in the NAc. Second, nicotine modulates nicotinic activity in the target areas of VTA, including the NAc. nAChR activity located at the dopaminergic terminals in the NAc augments DA release. While nAChRs containing the α2subunit are highly important for DA release in the striatum the α7subunit containing nAChRs also may have some role since NAc. In addition, nicotine enhances the contrast between tonic and phasic DA signals in the NAc by suppressing DA release resulting from tonic presynaptic inputs, while enhancing DA release from phasic burst-like presynaptic activity. Potential functional roles of nicotine-induced DA release in the NAc have been addressed in a recent review (Subramaniyan and Dani, 2015).
3.1.3 Hippocampus
the hippocampus may process contextual drug associations that contribute to context-evoked craving and drug-seeking behavior. For instant, Inactivation of the hippocampus with tetrodotoxin prevented context-stimulated reinstatement of cocaine-seeking behavior (Fuchs et al., 2005). Additionally, the role of the hippocampus in strengthening synaptic connections in efferent areas that are also involved in addiction
34
suggest that drug-induced changes in hippocampal function could produce long lasting functional changes in these areas (Kelley, 2004). These properties of the hippocampus make the hippocampus an intriguing area to study in regard to nicotine addiction.
On the other hand, α7 and α4β2nAChR subtypes have properties that could contribute to cellular changes associated with learning and addiction. Both subtypes are located in the hippocampus, and α4β2 and α7nAChRs are expressed pre and post-synaptically, suggesting that these receptor subtypes could modulate both pre and postsynaptic processes involved in synaptic plasticity (Gould, 2006). Furthermore, α4β2 and α7nAChRs are calcium-permeable, which could enhance activation of second messengers involved in synaptic plasticity (Broide and Leslie, 1999; Perry et al., 2002). These data suggest that nicotine binding at these nAChR subtypes may differentially affect learning processes in hippocampus (Gould, 2006).
3.1.4 Dopamine Receptor
G protein-coupled dopamine receptors (D1, D2, D3, D4, and D5) mediates all of the physiological functions of the catecholaminergic neurotransmitter dopamine, managing from voluntary movement and reward to hormonal regulation and hypertension (Beaulieu and Gainetdinov, 2011). The physiological actions of dopamine are mediated by five distinct but closely related G protein-coupled receptors (GPCRs) that are divided into two kind major classes, the D1 and D2 groups of dopamine receptors (Andersen et al., 1990). This classification is generally based on the basic biochemical observations revealing that dopamine can modulate adenylyl cyclase (AC) activity (Boyd and Mailman, 2012). Each members of the subfamilies of the D1- and D2-class receptors share a high level of homology of their transmembrane domains and have distinct pharmacological properties. It is commonly accepted that the D1-class dopamine receptors (D1 and D5) activate the
35
Gαs/olf family of G proteins to stimulate cAMP production by AC and are found exclusively
postsynaptically on dopamine-receptive cells, such as GABA-ergic medium spiny neurons (MSNs) in the striatum.
The D2-class dopamine receptors (D2, D3, and D4) couple to the Gαi/o family of G
proteins and thus induce inhibition of AC. In contrast to the D1-class dopamine receptors, D2 and D3 dopamine receptors are expressed both postsynaptically on dopamine target cells and presynaptically on dopaminergic neurons (Rankin, 2005; Rondou, Haegeman and Van Craenenbroeck, 2010). The dopamine D2 receptor is a dopamine receptor which is most frequently studied due to distribute in throughout of central nerve system, D2 receptors mediate an array of fundamental brain functions, including reward behavior, regulation of movement, learning and memory, and attention (Brancato et al., 2014; Puig et al., 2014).
3.1.5 Dopamine Signal on Nicotine Dependence
Nicotine also increases the firing rate of midbrain dopamine neurons by stimulation of α4β2nAChRs to promote nicotine dependence via dopamine receptor (Hsu et al., 1996; McCallum et al., 2006; De Biasi and Dani, 2011). Dopamine receptors are classified into two subfamilies, D1-like (D1 and D5) and D2-like (D2, D3, and D4)(Gingrich and Caron, 1993; C Missale et al., 1998). In neuronal coculture from coronal rats brain slices containing either NAc or VTA, nicotine stimulation increases CREB gene expression via stimulation both D2R and D1R (Inoue et al., 2007). Both receptor stimulation activate cAMP and Ca2+ signaling through Gαs/olf in D1R and Gi-stimulated Gβγ function in D2R, respectively
(Baik, 2013). D1R on NAc facilitates drug reward,exclusive genetic enhancement of D1R gene in either NAc core or NAc shell in mice (Gore and Zweifel, 2013). Additionally, stimulation of D1R on postnatal NAc promotes psychostimulant-induced behavior
36
sensitization via activation of PKA pathways, which enhance phosphorylation levels of GluA1 in mice brain (Chao, Ariano, et al., 2002; Chao, Lu, et al., 2002; Mangiavacchi and Wolf, 2004). The pivotal role of D1R on nicotine dependence is confirmed because administration of D1R antagonist SCH-23390 in NAc, ACC and PtA inhibits nicotine-induced behavior including dependence in mice (Kutlu et al., 2013; Hall et al., 2015a). Nicotine stimulates D1R to enhance CREB binding to CRE element (DynCREs) of the PD promoter, preferentially DynCRE3 inhibits by SCH23390 administration (McCarthy
et al., 2012). Thus, pivotal role of D1R has been well documented.
However, D2R functions in nicotine dependence remain unclear. There are a few evidences were reported. For example, chronic nicotine administration in a rat elevates the dopamine D2 high receptor level (Novak, Seeman and Foll, 2010). In vitro study using GST-fusion protein containing IL3 (GST-D2R-IL3), D2R binds CaMKIIα via the D2R IL3 domain(S. F. Zhang et al., 2014)thereby affecting long term memory of nicotine dependence because CaMKII is essential for learning formation particularly in CA1 region (Elgersma, 2004; Irvine et al., 2006; Lisman, Schulman and Cline, 2012). D2R have been inviolable effect in reward processing of drugs and natural stimulus such as food, D2R mediate approach-avoidance tendencies in smokers (Zlomuzica et al., 2018). Takeuchi et
al. shows on the NGD2L cell, quinpirole a D2R agonist enhances the Ca2+ intracellular level
then activates nuclear isoform of (CaMKII) δ3 increased exon-4 BDNF gene expression through D2LR, furthermore the isoform of (CaMKII)δ3 in rat subtantia nigra involved on modulates the exon-2 BDNF gene expression through CREB phosphorylation pathway (Takeuchi, Fukunaga and Miyamoto, 2002; Kamata, Takeuchi and Fukunaga, 2006). Therefore, the elucidation of mechanism of D2R-mediated nicotine dependence is important.
37
A recent study shows BDNF expression in the brain is managed by the serotoninergic and the dopaminergic neurotransmitters which are involved in nicotine addictive behavior (Mössner et al., 2000; Guillin et al., 2001; Seth et al., 2002). In the serotoninergic system, BDNF treatment on B lymphocyte 5-HTTLPR cell culture dose-dependent reduces the uptake and proliferation of (5-HT)serotonin (Mössner et al., 2000), BDNF from dopaminergic neuron is critical for D3R expression on NAc as a part on substance addicted (Guillin et al., 2001). Additionally, hippocampus BDNF was regulated by various neurotransmitters which are sensitive to nicotine administration and probably to become the critical molecule target of nicotine on modulates behavior (Zafra et al., 1990; Berninger et al., 1995; Vaidya et al., 1997). On Sprague Dawley rats the administration of serotonin agonist reduces the BDNF mRNA level on hippocampus (Vaidya et al., 1997), as evidence the serotoninergic system interacts with BDNF. Stimulation of GABAA receptor
activates the voltage gate Ca2+ in immature hippocampus culture increase the BDNF mRNA
level on gabaergic system (Berninger et al., 1995). Further study shows the BDNF on rat hippocampus is upregulated by the activation of muscarinic receptors and correlated with the acetylcholine release (Knipper et al., 1994).
3.2 Objectives
In the present study, we focus the role of dopamine D2R signal and BDNF expression in nicotine dependence using D2RKO mice. We describe the mechanism underlying D2R-mediated nicotine dependence in mice. These results suggest that D2 receptor is target for therapeutics of nicotine dependence.
38
3.3 Materials and Methods 3.3.1 Animals
Male C57BL/6JJmsSIC mice as WT mice aged 8 weeks (20–30 g) purchased from SLC (Hamamatsu, Japan), and male D2RKO mice aged 8 weeks (20-30 g) obtained from the Laboratory of Pharmacology, School of Pharmacy, Tohoku university were used in all experiments. The following protocol was used to generate D2RKO mice. A mutation was generated in the DA D2 receptor gene using homologous recombination in embryonic stem
cells and a targeting vector to delete the entire exon 7 and the 5′ half of exon 8, which is the region encoding the majority of the putative third intracellular loop, the last two transmembrane domains, and the carboxy terminus. Blastocyst injection was used to generate chimeric mice of the heterogenous 129/Sv × C57BL/6J background. F1 heterozygous mice sired by the chimeras were interbred to generate F2 mice of the
genotypes D2‐/‐. Backcrossing F2 mice to the C57BL/6J mouse strain for five generations
resulted in the incipient congenic N5 mice of D2‐/‐ genotypes used here. The genotype of the
mice was confirmed using PCR (Kelly et al., 1997). Mice were housed in a room with a 12/12-hour light/dark cycle (lights on at 09:00). Room conditions were temperature controlled at 22.0 ± 2 °C with a relative humidity of 55% ± 5%. Mice had free access to food and water. All experimental animal procedures were approved by the Committee on Animal Experiments at Tohoku University, and studies were conducted in accordance with committee guidelines. Every effort was made to minimize suffering and limit the number of animals used.
39
3.3.2 Experimental design
3.3.2.1 Induction of CPP by nicotine
Two groups of male mice (C57BL/6JJmsSIC) were treated using saline 0.9% i.p (n=6) or nicotine 0.5mg/kg i.p (n=8) All mice were acclimatized to the CPP box for 5 days after which, the preconditioning test was performed. Further, treatment was administered to the mice 28 days after the preconditioning. Briefly, mice in the vehicle group received a saline injection (i.p.) and were confined to the designated compartment for up to 30 minutes per day. Mice in the nicotine group received a nicotine injection (0.5 mg/kg, i.p.) followed by placement in the nicotine-paired compartment for 30 minutes. The CPP test was performed twice on day 15and day 29 after the conditioning phase. The place preference score of each mouse was recorded to determine reinforcing effect of nicotine on CPP. Thereafter, the mice were sacrificed on day 29, and the NAc and CA1 regions were dissected out from the mice brains; immunoblotting was performed to examine the D1R (n=5) and D2R (n=5) expression levels.
3.3.2.2 D1 and D2 receptor antagonist alleviates nicotine-induced CPP
WT mice were divided into 4 groups according to the treatment administered: saline 0.9% i.p (n=6), nicotine 0.5 mg/kg i.p (n=7), nicotine 0.5 mg/kg i.p+SCH23390 0.03 mg/kg s.c (n=6), and nicotine 0.5 mg i.p+Eticlopride-HCl 0.03 mg/kg (n=7). Eticlopride-HCl and SCH23390 were administered subcutaneously 30 minutes before nicotine injection. The behavior test was performed following the protocol for nicotine induction of CPP in the above-mentioned experiment.
3.3.2.3 Nicotine CPP test for WT and D2RKO mice
Mice were divided to 4 groups: WT vehicle (n=9), WT nicotine (n=10), D2RKO vehicle (n=8), and D2RKO nicotine (n=8). Behavioral assessments were conducted using
40
CPP methods as described above. Mice were sacrificed after the preference score assessment on day 29; the NAc and CA1 regions were dissected out from the mice brains, and immunoblot analysis was performed to examine the level of CaMKII (n=6), ERK1/2
(n=6) ,PKCα (n=8), D1R (n=5), D2R (n=5), CREB (n=5), pro-BDNF (n=5), BDNF (n=5),
α4nAChRs (n=6), α7nAChRs (n=6). Immunostaining of pCaMKII (n=4) and pERK (n=4) was performed to support the immunoblotting results. The experimental protocols are shown in Fig. 15
Figure 15. Experimental design Nicotin-induced CPP. Scheme of the whole experiment.
Mice were habituated to the CPP apparatus for 5 days, followed by a preconditioning test to determine the nicotine-paired compartment. The preference test was performed twice after 14- and 28-day conditioning, and the mice were sacrificed after preference score conditioning assessment on day 29. Nucleus accumbens and CA1 regions were dissected and assessed using immunoblot analysis.
3.3.2.4 Drug administration
NaCl (Wako) 0.9% was dissolved in double-distilled water, and nicotine hydrogen tartrate 0.5 mg/kg (Sigma Aldrich), Eticlopride-HCl 0.03 mg/kg (Sigma Aldrich), and SCH23390 0.03 mg/kg (Sigma Aldrich) were dissolved in saline solution. Mice were given nicotine or saline intraperitoneally once daily for 28 consecutive days. Eticlopride-HCl and SCH23390 were administered subcutaneously 30 minutes before nicotine injection.
Day 0
Acclimatization Conditioning I (2 weeks) Preconditioning test Day 5 Conditioning test 1 Day 19 Conditioning test 2 Day 34 Nicotine 0.5mgkg-1 Dissection
Conditioning II (2 weeks) 1. Immunoblotting Day 35
Nicotine 0.5mgkg-1
41
3.3.2.5 CPP test
The CPP test was performed using a biased design with two phases: habituation (5 days) and conditioning (28 days)
3.3.2.6 Acclimatization
Acclimatization was performed for five days prior to preconditioning. This process was designed to remove any environmental stress and included the weighing and testing rooms, CPP apparatus, and drug administration.
3.3.2.7 Preconditioning test
The preconditioning test was used to determine which compartment should be drug-or nicotine-paired. The compartment with the lower ratio preference was used (Carboni and Vacca, 2002). One day after habituation, each mouse was placed in the grey compartment for 5 minutes with the guillotine door closed. Following this, the guillotine door was opened, and the mouse could access all compartments for 15 minutes. Time spent in each compartment was recorded, and the preference ratio was calculated using the following formula:
𝑃𝑟𝑒𝑓𝑒𝑟𝑒𝑛𝑐𝑒 𝑟𝑎𝑡𝑖𝑜 = 𝑆𝑜𝑗𝑜𝑢𝑟𝑛 𝑡𝑖𝑚𝑒 𝑖𝑛 𝑟𝑒𝑙𝑎𝑡𝑒𝑑 𝑐𝑜𝑚𝑝𝑎𝑟𝑡𝑚𝑒𝑛𝑡 (𝑠)
𝑇𝑜𝑡𝑎𝑙 𝑡𝑖𝑚𝑒 𝑠𝑝𝑒𝑛𝑡 𝑖𝑛 𝑎𝑙𝑙 𝑐𝑜𝑚𝑝𝑎𝑟𝑡𝑚𝑒𝑛𝑡𝑠 (𝑠) × 100%
3.3.2.8 Conditioning
Mice were conditioned for 28 days. Each day, they were injected with nicotine followed by confinement in the designated compartment. The nicotine compartment was paired with the least preferred side of the compartment (Carboni and Vacca, 2002). Four hours later, the same procedure was repeated with saline administration for all groups. Mice were then confined to the opposite compartment of the apparatus. The next day, the conditioning preference ratio was calculated to assess nicotine dependence. On days 15 and