1
Genetic Factors Associated with Congenital Anomalies of the Kidney and Urinary Tract Associated and a Spectrum
of Extrarenal Disorders
Kyoji Fukushima, Keisuke Sugimoto, Kohei Miyazaki, Tomoki Miyazawa, Takuji Enya, Hitomi Nishi, Mitsuru Okada, and Tsukasa Takemura
Department of Pediatrics, Kindai University Faculty of Medicine, Osaka-Sayama, Japan
Abstract
Background. Several genes, including those of the renin-angiotensin system (RAS), act in con- cert to guide mammalian renal development from early nephrogenesis to definitive nephron for- mation. Aberrant forms of genes involved in nephrogenesis alter kidney development, leading to congenital anomalies of the kidney and urinary tract.
Methods. We analyzed the main genes involved in nephrogenesis, PAX2, RET and RAS-related genes, in 17 children with renal dysmorphism.
Results. A total of 17 cases were reviewed;
pathogenic variants were identified in four and incidental variants in three. Two of three patients with PAX2 abnormalities manifested renal colo- boma syndrome. No eye lesion was present in the third, who exhibited a frame-shift mutation from heterozygous insertion of T in exon 11; facial and skeletal abnormalities were detected. In analysis of RAS-related genes, an angiotensinogen gene (AGT) mutation (M268T) was identified in a pa-
tient exhibiting dwarf kidney, facial and skeletal abnormalities, mental retardation, and pituitary hyperplasia. Another patient exhibited an angio- tensin II receptor type 1 (AGTR1) gene mutation (p.L191L). A renal specimen showed tubular dys- genesis; extrarenal abnormalities included skull ossification defects. RET abnormality was de- tected in two patients with heterozygous muta- tions. One exhibited Hirschsprung disease and right renal agenesis. The other exhibited oligome- ganephronia and hypothyroidism but not multi- ple endocrine neoplasia nor medullary thyroid carcinoma (MTC); the etiology of these abnor- malities is unclear, as is the patient’s risk for MTC. Follow-up is necessary.
Conclusions. Gene aberration was detected in seven of 17 patients (41%). Unidentified genetic causes may contribute to pathophysiology in the remaining 10 patients.
Key words:gene mutation, renal dysmorphism, extrarenal abnormalities
Introduction
Most of the kidney in adult mammals develops from the metanephros, which begins differentia- tion upon extension of the ureteric bud (UB) from the caudal Wolffian duct to the dorsal metaneph- ric mesenchyme 1. During nephrogenesis, a net- work of several genes, including those that en- code the renin-angiotensin system (RAS), direct branching of the UB along with the formation and action of the mature nephrons 2. Aberrant forms
Received August 17, 2017; Accepted October 26, 2017
of genes involved in nephrogenesis can alter kid- ney development, leading to congenital anoma- lies of the kidney and urinary tract (CAKUT).
CAKUT is a spectrum of structural renal malfor- mations, including hydronephrosis, renal agene- sis, renal hypodysplasia, multicystic dysplastic kidney, ureteropelvic junction obstruction, vesicoureteral reflux (VUR), ureter duplex, megaureter, and posterior urethral valves 3. CAKUT can lead to chronic renal failure in chil- dren, and the prevalence of these malformations
has been estimated at 3–7 in 1000 births 4. CAKUT exhibits a range of severity, from mild hydronephrosis to unilateral renal agenesis. No- tably, the etiology of CAKUT remains unknown.
The development of CAKUT is associated with environmental factors and exposures, as well as some genetic factors related to nephrogenesis.
Aberrant forms of the RET and GDNF genes cause severe urinary tract abnormalities, includ- ing renal agenesis 5, while aberrant forms of the PAX2, HNF1β, SALL1, and RAS genes mainly cause hypoplasia and dysplasia 6-8.
The PAX2 gene, a member of the PAX family, controls other genes through directed synthesis of the protein paired box and alterations in DNA binding 9. During the embryonic stage, PAX2 is involved in development of the eyes, ears, central nervous system, and urogenital system. PAX2 ab- normality may result in renal coloboma syndrome (RCS). PAX2 is expressed in the mesoderm, caus- ing formation of the metanephros, Wolffian duct, and UB during nephrogenesis 10. When PAX2 ex- pression is reduced, the number of nephrons de- creases and subsequent renal hypoplasia occurs.
Abnormality of PAX2 also impairs myelination in visual, auditory and central nervous system struc- tures, resulting in related morphologic and func- tional abnormalities 10. The RAS genes are im- portant in maintaining physiological functions, including regulation of blood pressure and bal- ance of electrolytes; these genes also contribute to organogenesis, including formation of the met- anephric kidney 11,12. RAS gene aberrations in- duce diverse clinical phenotypes, including ab- normal renal tubule formation, ureteropelvic junction stenosis, megaloureter, multicystic dys- plastic kidney, and posterior urethral valves 6. HNF1β is a transcription factor that has been linked to proximal tubular differentiation in mouse studies 13. HNF1β mutations in humans are associated with a broad spectrum of diseases, in- cluding renal cysts, diabetes, and maturity onset diabetes of the young–type 5, as well as with he- patic, genital, and pancreatic abnormalities that lead to a variety of renal and extrarenal manifes- tations 14. SALL1 functions in anal, limb, and ear development as well as renal development; its mutations cause Townes-Brock syndrome 15.
A prior study used targeted exome sequencing to screen 122 CAKUT patients and discovered that 5% of these patients harbored deleterious rare variants or novel mutations in the GDNF-
CAKUT are associated with a wide range of ex- trarenal complications; thus, genetic analysis can aid in understanding the pathophysiology of CAKUT. Our purpose in this study was to use a genetic approach to identify the etiology of CAKUT in patients. We analyzed PAX2, RET and multiple RAS genes (angiotensinogen, angi- otensin II (Ang II), and Ang II receptor types 1 and 2) in 17 pediatric patients with abnormal re- nal formation who were referred to our depart- ment.
Materials and Methods Subjects
Our subjects included 17 patients with congen- ital renal dysplasia who were referred to our de- partment.
Methods
Genomic DNA extraction, polymerase chain reaction (PCR), and determination of gene se- quences
Approximately 5 mL of peripheral blood was collected from patients into tubes containing Na- EDTA; genomic DNA was extracted from the blood using NucleoSpin for Blood (TaKaRa Bio Inc, Shiga, Japan). Human genomic DNA (Clon- tech, code 636401, CA) was used as a control. Pa- tient samples and control genomic DNA were di- luted with sterile water to prepare 10 ng/μL solu- tions. PCR was performed using the diluted DNA solutions as templates; the reaction was per- formed in a PCR Thermal Cycler Dice Gradient (TaKaRa Bio Inc, Shiga, Japan). To determine the extent of deletions and to identify break points, PCR primers were designed to amplify fragments of approximately 200–300 bp, based on gene se- quences registered in GenBank. Primers for am- plifying PAX2 exon 11 were 5’- ATGTCTCCTCACCCGTGGATC-3’ (forward, F) and 5’-AGGCCCAGGCCTAACCTGCTAAA- 3’ (reverse, R); angiotensinogen (AGT) primers were 5’-GATCTGGTTAGATGGCACTTA-3’
(F), and 5’-AAAGGTGGGAGACTGGGGGTG-3’
(R); angiotensin receptor 1 (AGTR1) primers were 5’-GTTACTACGTTTATGACTGAG-3’ (F), and 5’-CCACATAATGCATTTTGTCCT-3’ (R);
angiotensin receptor 2 gene (AGTR2) primers were 5’-CTAATGATTCAAGGATGTCCT-3’ (F), and 5’-GATTCAGAAAAGATTAGGGGA-3’
(R); RET exon4 primers were 5’-CACAG-
5’-
GAGGTGATCCGCCTTCCCGTCCTTCTCCAA -3’ (R). For PCR, the amplification protocol for PAX2 was 94℃ for 30 seconds, 64℃ for 30 sec- onds (annealing), and 72℃ for 1 minute (exten- sion), repeated for 30 cycles; the protocol for AGT, AGTR1 and AGTR2 was 94℃ for 30 sec- onds, 64℃ for 30 seconds, and 72℃ for 30 sec- onds, repeated for 35 cycles; and the protocol for RET was 98℃ for 10 seconds, 63℃ for 15 sec- onds, and 68℃ for 1 minute, repeated for 30 cy- cles.
Direct sequencing
PCR products were enzymatically purified and templates for sequencing were prepared. The se- quencing reaction was performed using the DNA templates and the dye terminator method of the BigDye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA). Reaction prod- ucts were purified by gel filtration, and sequence analysis was performed with a capillary-type se- quencer, ABI3730xl (Applied Biosystems, CA, USA).
Results
Gene aberrations were detected in four of the 17 patients (PAX2, 3; RET, 1); incidental variants were detected in three patients (RAS genes, 2;
RET, 1). Clinical features and gene mutations in these seven patients are summarized in Table 1.
Unreported 5 cases are described below.
1. PAX2 gene aberration
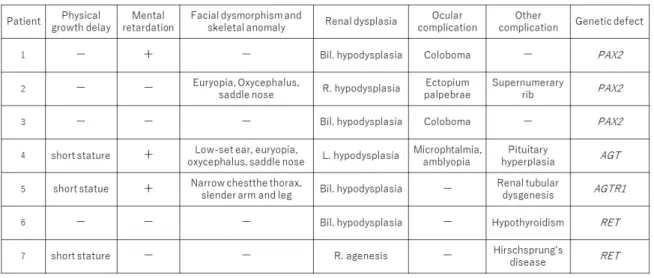
Patient 2 exhibited occult blood in the urine at 7 years of age. Imaging revealed right renal hy- poplasia (Figure 1a). Extrarenal abnormalities in- cluded characteristic facial features (acrocephaly, hypertelorism, ectropium palpebrae spasticum, and saddle nose; Figure 1b) and a supernumerary rib (Figure 1c). Visual acuity and fields were nor- mal; no abnormalities were detected in the retina or optic nerve. Family history was noncontribu- tory. His most recent (15 years of age) creatinine clearance rate (Ccr) was 58.4 mL/min/1.73m2. Gene analysis identified a heterozygous frame- shift mutation, with an insertion of T in exon 11 (nucleotide or NT position, 21 336 271-21 336 272; Figure 1d). The left kidney appeared normal by imaging, but abnormally few glomeruli (3.6/mm2), which were enlarged, were evident in a renal biopsy specimen; some were segmentally sclerotic (Figure 1e).
Patient 3 exhibited proteinuria at 6 years of age.
Imaging revealed bilateral renal hypoplasia (Fig- ure 2a). Extrarenal symptoms include strabismus, first noted at 2 years; at that time, she was diag- nosed with optic nerve hypoplasia. Her father ex- hibited end-stage renal disease and visual impair- ment of unknown cause. Her most recent Ccr (14 years of age) was 38 mL/min/1.73m2, demonstrat- ing progression of renal insufficiency. Gene anal- ysis identified a heterozygous frame-shift muta- tion, with C inserted in exon 2 (NT position, 21 258 147-21 258 148; Figure 2b). A similar muta- tion was identified in her father.
Table 1 Clinical features and mutated genes in patients with renal dysplasia
Figure 1 Patient 2. Images, facial features, and histologic findings of the kidney (periodic acid-Schiff stain, original magnification, x40), and gene analysis findings.
The right kidney is hypoplastic (CT in a, indicated by circle), and a supernumerary right rib (radiograph in c, indicated by arrow) is present. Facial features are abnormal (acrocephaly, hypertelorism, ectropium palpebrae, and saddle nose; b). On histologic examination of the kidney, glomeruli are few (3.6 /mm2) and enlarged; with some exhibiting segmental sclerosis (e). On PAX2 gene analysis, a frame-shift mutation caused by heterozygous insertion of thymine (T) is detected in exon 11 (d, indicated by circle).
Figure 2 Abdominal CT findings and PAX2 gene analysis in Patient 3.
Both kidneys are hypoplastic (a). Gene analysis shows a frame-shift mutation involving heterozygous insertion of cytosine (C) in exon 2 (b, indicated by circle).
2. RAS gene aberration
Patient 4 was born at 39 weeks and 2 days of gestation; birth height and weight were 44 cm and 2204 g. He exhibited unusual facial features when he was 1 year old. He also exhibited a left dwarf kidney with small cysts (Figure 3a). Extrarenal abnormalities included unusual facial features (acrocephaly, microphthalmia, saddle nose, and low-set ears; Figure 3b); mental retardation (IQ, 42); pituitary hyperplasia; and foramen magnum stenosis (Figure 3c). His abdomen was flat, upper and lower limbs were slender and elongated, stat- ure was small (15 years of age): height, 148 cm (-
history was noncontributory. Current renal func- tion was stable. Gene analysis identified a homo- zygous mutation of M268T (T/T→C/C) in exon 2 of AGT (Figure 3d).
Patient 5 was born at 37 weeks and 1 day of gestation; birth weight was 2374 g. Tachypnea and cyanosis were evident after birth, and no uri- nation occurred during the first 24 hours of life.
Renal function abnormalities and bilateral dwarf kidneys were detected (Figure 4a) when he was referred to our department. Notably, his mother was not prescribed angiotensin inhibitors or re- ceptor antagonists during pregnancy. His present
body weight was 10.9 kg (-2.6 SD). Extrarenal abnormalities included mental retardation and skeletal defects such as a small abdomen, slender upper and lower limbs, and areas of skull thinning (Figure 4b). Urinalysis revealed low specific gravity and excessive urinary β2-microglobulin
(19420 μg/L). A renal biopsy specimen-obtained at 3 years of age-showed marked cystic dilation of renal tubules, mainly in distal tubules (Figure 4c). Gene analysis identified a heterozygous mu- tation, p.L191L (C/C→T/C), in exon 5 of AGTR1 (Figure 4d).
Figure 3 Patient 4. CT findings, facial features, renal histologic findings, and AGT gene analysis.
The left kidney is hypoplastic (a). Abnormal facial feature such as acrocephaly, microphthalmia, saddle nose, and low-set ears, are noted (b). In magnetic resonance imaging of the head (c), pituitary hyperplasia (indicated by circle) and foramen magnum stenosis (indicated by arrow) are evident. Gene analysis (d) shows homozygous mutation of M268T in exon 2 in the patient, while the father and mother each show a heterozygous G/G mutation.
Figure 4 Patient 5. Imaging findings of the kidneys (a, b), renal histologic findings (c), and AGTR1 gene analysis (d).
Bilateral dwarf kidneys are seen to contain small cysts (a). The left kidney shows dilation of the renal pelvis.
Ossification of the skull is partially defective (b, indicated by arrow). On histologic examination of the kidney, renal tubules are markedly dilated; findings are suggestive of renal tubular dysgenesis (c). AGTR1 gene analysis discloses a heterozygous mutation, p.L191L, in exon 5 (d).
3. RET gene aberration
Patient 6 had no adverse perinatal events and a normal birth weight. Renal dysfunction was de- tected at 1 year of age (blood urea nitrogen (BUN), 24 mg/dL; serum creatinine (s-Cr), 0.6 mg/dL).
Increased serum thyroid-stimulating hormone (TSH; 12.3 mU/mL) was noted, but free thyroid hormone concentrations (FT3, FT4) were normal.
No auto-antibodies, including anti-thyroid recep- tor or anti-thyroid peroxidase antibodies, were detected. Currently, (23 years of age), TSH was normalized because of thyroxine administration.
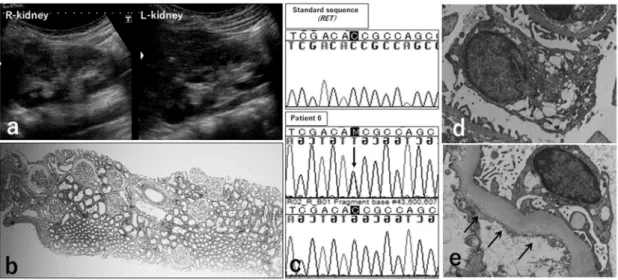
No imaging or laboratory findings have suggested medullary thyroid cancer (MTC). Renal ultraso- nography at 21 years of age showed both kidneys to be small. Irregular parenchymal echoes were seen; overall echogenicity was increased (Figure 5a). Ccr was 61.3 mL/min/1.73m2, indicating mild renal insufficiency. On histologic examina- tion of a renal biopsy specimen, there were few glomeruli (5/mm2), which were enlarged (oligo- meganephronia; Figure 5b). Gene analysis identi- fied a heterozygous mutation of T278N (C/C→C/A) in exon 4 (Figure 5c).
Figure 5 Patient 6. Renal imaging findings, renal histological findings, and RET gene analysis.
The right kidney shows dwarf proportions and renal parenchymal echogenicity is increased (a). Histologic ex- amination of the kidney shows reduced numbers of glomeruli (5/mm2) and glomerular enlargement (oligoneph- ronia; b). Gene analysis shows a heterozygous mutation, T278N in exon 4 (c, arrow). On electron microscopy, enlargement of podocytes with increased cytoplasmic vacuolization (d, magnification, x15,000) and thickening of the glomerular basement membrane (e, indicated by arrow, magnification, x25,000).
Discussion
PAX2 encodes a critical transcription factor that is expressed in the nephron progenitors 17. PAX2 mutations are most commonly associated with RCS (also called papillorenal syndrome), an autosomal dominant disorder that is characterized by optic nerve malformations (optic nerve colo- boma, optic nerve dysplasia) and renal defects (oligomeganephronia, hypodysplasia with or without VUR, and renal cysts) 18. Other, less com- mon extrarenal manifestations include sensori- neural hearing loss and brain malformations 19. PAX2 gene aberration was detected in three pa- tients in our study. Patient 1, who was previously described 20, manifested P130H as a novel muta- tion; however, most clinical features of RCS were present. Patient 3 also developed common clini-
inherited case with a frame-shift mutation in- duced by heterozygous insertion of cytosine (C) in exon 2. PAX2 abnormalities associated with RCS include five types of frame-shift mutation in exons 2 or 5; two types of missense mutations in exon 3; and observation of chromosomal translo- cation of PAX 2 introns; among these, 619insG in exon 2 has been reported most frequently 21. The frame-shift mutation in exon 2 (in our Patient 3) has not been reported previously. On the other hand, no ocular abnormalities were observed in Patient 2, but skeletal malformations were present, including abnormal facial features and a supernu- merary rib. During prenatal development, PAX2 first is expressed in the future midbrain and hind- brain, where it acts in migration and proliferation of cranial neural crest (CNC) cells 22. PAX2 also
structure by promoting differentiation of CNC- derived tissues 23. A frame-shift mutation from heterozygous insertion of thymidine (T) into exon 11 was detected in Patient 2, where the abnormal differentiation of CNC-derived tissues may have caused facial dysmorphism and skeletal abnor- malities instead of the typical RCS phenotype as- sociated with PAX2 gene aberrations. The degree of optic nerve coloboma varies, both between pa- tients and between each eye in an individual, even in patients with identical PAX2 mutations 24. Moreover, a recent study revealed that identical twins with PAX2 mutations exhibited different eye and kidney abnormalities 25. Consistent with our study, point mutations within the coding re- gion of PAX2 may not represent the only cause of RCS 26. Analysis of the PAX2 gene may be indi- cated in patients with both facial dysmorphisms and urinary tract abnormalities. PAX2 gene aber- rations may be associated with oligomeganeph- ronia 27, as shown in renal biopsy specimens.
Taken together, these findings indicate that mul- tiple factors may exert additive effects on the pro- gression of kidney and eye dysfunctions.
The RAS system plays a critical role in blood pressure and fluid/electrolyte homeostasis. In the kidney, RAS components are expressed in both temporal- and spatial-dependent manners during UB branching and nephron formation; notably, Ang II participates in nephron formation 12. In Pa- tient 4, we observed hypoplasia of the left kidney, mental retardation, and pituitary hyperplasia. The causal relationship between the AGT gene abnor- mality and the pituitary defect in this child is un- clear; however, RAS genes play a role in mainte- nance of cerebral microvascular circulation and differentiation, as well as maturation of nerve cells 11. Yosypiv et al. reported large low-set ears, limb-position defects, arthrogryposis, and skull ossification defects as extrarenal consequences of AGT gene aberration 12. Ang II is involved in bone metabolism 28; importantly, abnormality of the AGT gene encoding the precursor of Ang II in Patient 4 would likely result in synthesis of an in- complete form of Ang II, which may be responsi- ble for the patient’s skeletal abnormalities. In Pa- tient 5, renal biopsy showed that renal tubules were markedly dilated, consistent with renal tub- ular dysgenesis (RTD). RTD is characterized by anuria immediately after birth due to hypoplasia of proximal tubules; skull ossification defects are also common. Early death from pulmonary hypo- plasia is likely 29. RTD inheritance is autosomal
dominant and involves various abnormalities of RAS genes 30. In Patient 5, anuria and tachypnea were noted immediately after birth; however, the survival of the patient suggests that some func- tional activity remains in the defective mutant protein encoded by AGTR1. At present, this pa- tient and Patient 4 exhibit similar mental retarda- tion phenotypes. Further, Takeshita et al. reported mental retardation associated with AGTR2 gene aberration, suggesting that the RAS system is im- portant for neuronal development 31. Since the AGTR1 mutation in patient 5 may serve as a syn- onymous substitution, the exact cause of the neu- ronal defects is still unknown. However, Patient 5 exhibited RTD and skeletal abnormalities char- acterized by gene mutations in the RAS system.
Other genes such as REN (renin), which causes RTD, may be involved.
The RET gene, located at q.11.2 of chromo- some 10, is a proto-oncogene encoding receptor tyrosine kinase 32. The RET protein, which com- plexes with glial cell-derived neurotrophic factor (GDNF) and its receptor, GFRα1, is involved in cell proliferation and differentiation in the kidney, nervous system, and genital organs 30. During kid- ney development, signal transduction through the GDNF-RET system is essential for sprouting of the UB, followed by branching of the UB through GDNF-RET interactions with several genes 33. RET mutations can result in either loss-of-func- tion or gain-of-function mutations 34. Heterozy- gous mice (GDNF+/-) exhibit significantly fewer nephrons that display structural changes on histo- logical analysis, such as enlargement of podo- cytes with increased cytoplasmic vacuolization and marked thickening of the glomerular base- ment membrane 35. Similar findings were also rec- orded in Patient 6, indicating that RET mutation may contribute to renal dysplasia. Additionally, loss of RET gene function is involved in develop- ment of the extrarenal pathology, Hirschsprung disease (HSCR) 36. Conversely, gain-of-function mutations are involved in multiple endocrine ne- oplasia (MEN) syndromes: MTC and pheochro- mocytoma 37. In Patient 6, renal dysplasia (loss- of-function) and hypothyroidism (possible gain- of-function) may have occurred simultaneously as a result of the heterozygous T278N mutation.
However, MTC did not develop. Since this muta- tion affects the extracellular cadherin-like do- main of RET, it may influence binding between GDNF and GFRα1. Epithelial cells in the thyroid follicle arise from thyroid stem cells; abnormal
rearrangement of the RET/PTC gene inhibits this differentiation process and can cause carcinogen- esis 38. The resulting incompletely differentiated follicular cells might exhibit impairment of thy- roid hormone secretion, with increased TSH re- quired for adequate secretion. MEN and MTC are possible consequences of gain-of-function muta- tions in the RET gene. However, one patient with an RET gene mutation showed hypothyroidism accompanying Hashimoto disease; this patient also exhibited cystic renal lesions 39. A patient with simultaneous MEN and HSCR also has been reported 40, implying the presence of a gene mu- tation that results in both loss and gain of function.
Patient 7, who was previously described 41, had a p.S811F mutation in exon 14. Reduction of RET gene expression to 30% of normal levels led to HSCR-like neural defects in the distal large intes- tine; however, no renal consequences were ob- served in a mouse model. In a RET(–/–) mouse model, neural elements in the distal large intes- tine were lost and marked renal hypoplasia was observed 42, suggesting that the RET mutation in our patient substantially interfered with a func- tioning protein product. On analysis of molecular structure, we found that a large phenylalanine side chain—resulting from substitution for ser- ine—most likely interfered with ATP processes that include sufficient cAMP production, result- ing in marked enzyme inactivation. Renal pathol- ogy is not sufficient to elucidate the mechanism of congenital development of a unilateral kidney or of lateral renal differences in patients with two kidneys. However, differences in gene expression may contribute to the severity of disease, includ- ing renal size differences. Furthermore, fusion at the embryonic stage, and the existence of VUR after birth, suggests kidney atrophy on the side of the lesion.
Conclusion
We investigated abnormalities in PAX2, RET and RAS genes, which are frequent causes of re- nal and urinary tract malformations. Gene aberra- tion was detected in seven of 17 patients. Since unidentified genetic causes might contribute to pathophysiology in the remaining 10 patients, ex- tensive gene analysis using whole-exon sequenc- ing may be necessary.
Authors' contributions
the Kindai University Graduate School, Faculty of Medicine. This manuscript is not under consid- eration for publication elsewhere, in any language, except as an abstract.
Acknowledgements
We thank Ai Itoh for technical support in man- uscript preparation. This study was partial sup- ported by a Grant-in-Aid for Scientific Research from Osaka Kidney Bank (2016) and from Mori- naga Hoshikai (2015 to 2016). We thank Ryan Chastain-Gross, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Informed Consent
The study was performed following approval by the Ethics Committee of Kindai University Faculty of Medicine (approval number 26-204).
Written informed consent was acquired from pa- tients or the patients’ parents/guardians. Use of renal specimens for evaluation of renal histologic findings, and for reproduction in figures, was ap- proved by patients or patients’ parents and/or guardians.
Conflict of interests
The authors declare that they have no compet- ing interests involving this work.
References
1 .Risdon RA (1992) Development of the kidney. In Heptinstall RH, editor. Pathology of the Kidney. Bos- ton: Little, Brown, pp93-101
2.Phua YL, Ho J (2016) Renal dysplasia in the neonate.
Curr Opin Pediatr 28: 209-215
3.Vivante A, et al. (2014) Single-gene causes of con- genital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol 29: 695-704 4.Livera LN, Brookfield DS, Egginton JA, Hawnaur JM
(1989) Antenatal ultrasonography to detect fetal renal abnormalities: a prospective screening programme.
BMJ 298: 1421-1423
5.Benz K, et al. (2011) Early glomerular alterations in genetically determined low nephron number. Am J Physiol Renal Physiol 300: 521-530
6.Song R, Yosypiv IV (2011) Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 26: 353-364
7.Hilliard SA, El-Dahr SS (2016) Epigenetics mecha-
1060
8.Nishinakamura R (2016) Stem cells and renal devel- opment in 2015: advances in generating and maintain- ing nephron progenitors. Nat Rev Nephrol 12: 67-68 9.Cheong HI, et al. (2007) A clinico-genetic study of
renal coloboma syndrome. Pediatr Nephrol 22: 1283- 1289
10.Sanyanusin P, et al. (1995) Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nature Genet 9:
358-364
11.Schütz S, Le Moullec JM, Corvol P, Gasc JM (1996) Early expression of all the components of the renin-an- giotensin-system in human development. Am J Pathol 149: 2067-2079
12.Yosypiv IV (2014) Renin-angiotensin system in ure- teric bud branching morphogenesis: implications for kidney disease. Pediatr Nephrol 29: 609-620
13.Paces-Fessy M, Fabre M, Lesaulnier C, Cereghini S (2012) Hnf1b and Pax2 cooperate to control different pathways in kidney and ureter morphogenesis. Hum Mol Genet 21: 3143-3155
14.Nakayama M, et al. (2010) HNF1B alterations asso- ciated with congenital anomalies of the kidney and uri- nary tract. Pediatr Nephrol 25: 1073-1079
15.Kohlhase J (2000) Sall1 mutations in Townes-Brocks syndrome and related disorders. Hum Genet 16: 460- 466
16.Chatterjee R, et al. (2012) Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling com- plex in a cohort of living US patients with urinary tract malformations. Hum Genet 131: 1725-1738
17.Gong KQ, Yallowitz AR, Sun H, Dressler GR, Wellik DM (2007) A Hox-Eya-Pax complex regulates early kidney developmental gene expression. Mol Cell Biol 27: 7661–7668
18 .Madariaga L, et al. (2013) Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol 8: 1179-1187 19.Schimmenti LA, et al. (1997) Further delineation of
renal-coloboma syndrome in patients with extreme var- iability of phenotype and identical PAX2 mutations.
Am J Hum Genet 60: 869-878
20.Miyazawa T, et al. (2009) A case of renal-coloboma syndrome associated with mental developmental delay exhibiting a novel PAX2 gene mutation. Clin Nephrol 72: 497-500
21.Stuart ET, Gruss P (1996) Developmental control gene in cell growth and differentiation. Growth Differ- ent 7: 405-412
22.Mansouri A, Hallonet M, Gruss P (1996) Pax genes and their roles in cell differentiation and development.
Curr Opin Cell Biol 8: 851-857
23.Rowitch DH, McMahon AP (1995) Pax-2 expression in the murine neural plate precedes and encompasses the expression domains of Wnt-1 and En-1. Mech Dev 52: 3-8
24.Okumura T, et al. (2015) Association of PAX2 and
Other Gene Mutations with the Clinical Manifestations of Renal Coloboma Syndrome. PLoS One 10:
e0142843
25.Iatropoulos P, et al. (2012) Discordant phenotype in monozygotic twins with renal coloboma syndrome and a PAX2 mutation. Pediatr Nephrol 27: 1989-1993 26.Schimmenti LA (2011) Renal coloboma syndrome.
European journal of human genetics: EJHG 19: 1207- 1212
27.Salomon R, et al. (2001) PAX2 mutations in oligo- meganephronia. Kidney Int 59: 457-462
28.Nakagami H, Osako MK, Morishita R (2013) Poten- tial effect of angiotensin II receptor blockade in adi- pose tissue and bone. Curr Pharm Des 19: 3049-3053 29.Lacoste M, et al. (2006) Renal tubular dysgenesis, a
not uncommon autosomal recessive disorder leading to oligohydramnios: Role of the Renin-Angiotensin sys- tem. J Am Soc Nephrol 17: 2253-2263
30.Gubler MC (2014) Renal tubular dysgenesis. Pediatr Nephrol 29: 51-59
31.Takeshita E, Nakagawa E, Nakatani K, Sasaki M, Goto Y (2012) Novel AGTR2 missense mutation in a Japanese boy with severe mental retardation, pervasive developmental disorder, and epilepsy. Brain Dev 34:
776-779
32.Ishizaka Y, et al. (1989) Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 4: 1519- 1521
33.Reidy KJ, Rosenblum ND (2009) Cell and molecular biology of kidney development. Semin Nephrol 29:
321-337
34.Edery P, Eng C, Munnich A, Lyonnet S (1997) RET in human development and oncogenesis. Bioessays 19:
389-395
35.Benz K, et al. (2011) Early glomerular alterations in genetically determined low nephron number. Am J Physiol Renal Physiol 300: 521-530
36.Lantieri F, Griseri P, Ceccherini I (2006) Molecular mechanisms of RET-induced Hirschsprung pathogene- sis. Ann Med 38: 11-19
37.Pasquali D, et al. (2012) Multiple endocrine neoplasia, the old and the new: a mini review. G Chir 33: 370-373 38.Cote GJ, Grubbs EG, Hofmann MC (2015) Thyroid C-cell biology and oncogenic transformation. Recent Results Cancer Res 204: 1-39
39.Bano G, et al. (2013) A complex endocrine conun- drum. Fam Cancer 12: 577-580
40.Quedas EP, et al. (2012) RET haplotype, not linked to the C620R activating mutation, associated with Hirschsprung disease in a novel MEN2 family. Clinics (Sao Paulo) 67 Suppl 1: 57-61.
41.Sugimoto K, et al. (2016) Heterozygous p.S811F RET gene mutation associated with renal agenesis, oligome- ganephronia and total colonic aganglionosis: a case re- port. BMC Nephrol 17: 146
42.Uesaka T, Nagashimada M, Yonemura S, Enomoto H (2008) Diminished Ret expression compromises neu- ronal survival in the colon and causes intestinal agan- glionosis in mice. J Clin Invest 118: 1890-1898