崇城大学学位論文
B 型肝炎ウイルス増殖抑制効果を有する
4′-C-cyano-2′-deoxynucleosides の体内動態特性の評価
令和 2 年度
橋本 麻衣
崇城大学学位論文
B 型肝炎ウイルス増殖抑制効果を有する
4′-C-cyano-2′-deoxynucleosides の体内動態特性の評価
2021 橋本 麻衣
Evaluation of pharmacokinetic properties of 4′-C-cyano-2′-deoxynucleosides with hepatitis B virus
replication inhibitory effect
Mai Hashimoto
Evaluation of pharmacokinetic properties of 4′-C-cyano-2′-deoxynucleosides with hepatitis B virus replication inhibitory effect
Mai Hashimoto
4′-C-cyano-2′-deoxynucleosides were found to have high antiviral activity against hepatitis B virus (HBV). 4′-C-cyano-2′-deoxyinosine (CdI) is an inosine nucleoside analog, whereas 4′-C-cyano-2′- deoxyguanosine (CdG) is a guanosine nucleoside analog. The IC50 values for HBV are 0.045 µM and 0.0004 µM, respectively. In particular, CdG has high anti-HBV activity comparable to entecavir (ETV) in vitro and in vivo. In recent years, guanosine nucleoside analogs such as the 4′- modified with cyano group have been developed for HBV treatment. However, information on the pharmacokinetics of these 4′-C-cyano-2′-deoxynucleosides, which are indispensable for new drug development, is lacking.
ADME of the drug revealed by pharmacokinetic study provides useful, necessary information for investigating the duration of drug efficacy, prediction of the onset of side effects, optimal formulation and appropriate administration schedule. Therefore, I investigated the pharmacokinetics of CdI and CdG in this study. The results obtained in this study are summarized as follows:
1. Pharmacokinetics of 4′-C-cyano-2′-deoxyinosine (CdI) in healthy rats
In order to quantify CdI in biological samples, I developed optimal liquid chromatography-mass spectrometry (LC/MS) conditions and pretreatment methods. In the pretreatment method, CdI could be extracted from biological samples with a high recovery rate by the solid phase extraction (SPE) method using Oasis® MCX. Pharmacokinetic studies of CdI in healthy rats showed that CdI has relatively good blood retention and bioavailability. The absorbed CdI distributed more to the liver than the kidney and was unlikely to be metabolized by hepatic cytochrome P450 (CYP). CdI excreted in the urine unchanged over time. Co-administration of CdI and ETV affects their pharmacokinetics, and caution is required when CdI is administered in combination with structural analogs such as ETV.
2. Pharmacokinetics of 4′-C-cyano-2′-deoxyguanosine (CdG) in healthy rats
CdG could also be extracted from the biological sample by the SPE method using Oasis® MCX and quantified with the same LC/MS conditions as CdI. Pharmacokinetic analysis of CdG administered to healthy rats revealed that the bioavailability of CdG was comparable to other nucleoside analogs. CdG disappeared rapidly from the blood and distributed more to the liver than the kidney, but hardly metabolized by hepatic CYP. Furthermore, the absorbed CdG excreted in the urine unchanged. Since CdG and ETV's co-administration affects their pharmacokinetics, it is necessary to avoid concurrent administration of a drug having a similar structure with CdG.
3. Pharmacokinetics of 4′-C-cyano-2′-deoxyguanosine (CdG) in liver disease model rats
To investigate the effect of liver damage on the pharmacokinetic characteristics of CdG, we examined the pharmacokinetics of CdG in Concanavalin A (Con A)-induced liver injury model rats. The pharmacokinetic parameters after intravenous administration of CdG in Con A-induced viral liver injury (VLI) model rats were not significantly different from healthy rats. However, when CdG was orally administered, Cmax and AUC decreased significantly, and the amount distributed to organs also reduced compared to healthy rats. These decreases in pharmacokinetic parameters strongly suggest that inhibited absorption might have occurred in the gastrointestinal tract after the oral administration of CdG in the Con A-induced VLI model rat. The residual food in the stomachs of the Con A-induced VLI model rat was 13 times heavier than that of healthy rats, suggesting the possibility that residual food inhibits the absorption of CdG. The results obtained in this study indicate that liver damage has a negligible effect on the pharmacokinetics of CdG.
4. Pharmacokinetics of 4′-C-cyano-2′-deoxyguanosine (CdG) in renal disease model rats To investigate the effect of renal dysfunction on the pharmacokinetics of CdG, we performed a pharmacokinetic analysis of CdG in CKD model rats. In CKD model rats, a decrease in blood clearance of CdG showed a significant correlation with an increase in plasma creatinine level. The blood concentration of CdG increased significantly, and the pharmacokinetic parameters also changed substantially after oral administration of CdG in CKD model rats. Renal dysfunction did not cause accumulation of CdG in the kidneys and liver. The cumulative excretion in urine 24 hours after CdG administration was not significantly different from that in healthy rats, and the blood CdG level was below the detection limit. Therefore, although the blood half-life of CdG was prolonged due to the decrease in renal function, the possibility of CdG accumulating in the body is low.
In conclusion, this study was the first to clarify the pharmacokinetic properties of CdI and CdG, which are nucleoside analogs with the 4′-modified with cyano group, and 4′-C-cyano-2′-deoxynucleosides.
The study showed that CdI and CdG have good oral bioavailability; our current results can be important primary data for the future development of CdG as a new therapeutic agent for HBV.
本論文で使用した略語一覧
ADV adefovir
AGP α1-acid glycoprotein
AUC area under the concentration-time curve BCS biopharmaceutical classification systems CKD chronic kidney disease
CYP cytochrome P450
CdG 4′-C-cyano-2′-deoxyguanosine CdI 4′-C-cyano-2′-deoxyinosine Cmax maximum drug concentration Con A concanavalin A
ETV entecavir
GC gas chromatography
HBV hepatitis B virus
HIV human immunodeficiency virus
HPLC high performance liquid chromatography HSA human serum albumin
IFN interferon
LAM lamivudine
LC/MS liquid chromatography-mass spectrometry MATE multidrug and toxin efflux extrusion protein NRTI nucleoside analogue reverse transcriptase inhibitor OAT organic anion transporter
OCT organic cation transporter PEG polyethyleneglycol
RI radioisotope
SPE solid phase extraction
t1/2 half-life
TAF tenofovir alafenamide TDF tenofovir disoproxil fumarate
TFV tenofovir
tmax time to reach peak plasma concentration VLI viral liver injury
本論文は,学術雑誌に掲載された次の論文を基礎とするものである.
(1) Pharmacokinetics studies of 4′-cyano-2′-deoxyguanosine, a potent inhibitor of the hepatitis B virus, in rats
J. Pharm. Pharmacol., 70, 6, 723–731 (2018).
Mai Hashimoto, Kazuaki Taguchi, Takako Ishiguro, Satoru Kohgo, Shuhei Imoto, Keishi Yamasaki, Hiroaki Mitsuya, Masaki Otagiri
(2) Pharmacokinetic properties of a novel inosine analog, 4′-cyano-2′-deoxyinosine, after oral administration in rats
PLoS One, 13, 6, e0198636 (2018).
Mai Hashimoto, Kazuaki Taguchi, Takako Ishiguro, Satoru Kohgo, Shuhei Imoto, Keishi Yamasaki, Hiroaki Mitsuya, Masaki Otagiri,
(3) Pharmacokinetic properties of orally administered 4′-cyano-2′-deoxyguanosine, a novel nucleoside analog inhibitor of the hepatitis B virus, in viral liver injury model rats Biol. Pharm. Bull., 43, 9, 1426-1429 (2020).
Mai Hashimoto, Kazuaki Taguchi, Shuhei Imoto, Keishi Yamasaki, Hiroaki Mitsuya, Masaki Otagiri
(4) Pharmacokinetics of 4′-cyano-2′-deoxyguanosine, a novel nucleoside analog inhibitor of the resistant hepatitis B virus, in a rat model of chronic kidney disease
J. Infect. Chemother., In press (2020)
Mai Hashimoto, Kazuaki Taguchi, Shuhei Imoto, Keishi Yamasaki, Hiroaki Mitsuya, Masaki Otagiri
目 次
第1章 緒論 ... 1
第2章 4′-C-cyano-2′-deoxyinosine(CdI)の健常ラットにおける 体内動態解析 ... 6
第1節 序 ... 6
第2節 結果 ... 6
2-1 LC/MSによるCdIの測定法の確立 ... 6
2-2 血漿中濃度推移 ... 9
2-3 臓器分布 ... 10
2-4 タンパク結合 ... 11
2-5 代謝 ... 11
2-6 尿中排泄 ... 12
2-7 CdIとETVの併用投与時の体内動態 ... 13
第3節 考察 ... 15
第4節 小括 ... 18
第3章 4′-C-cyano-2′-deoxguanosine(CdG)の健常ラットにおける 体内動態解析 ... 19
第1節 序 ... 19
第2節 結果 ... 19
2-1 LC/MSによるCdGの測定法の確立 ... 19
2-2 血漿中濃度推移 ... 21
2-3 臓器分布 ... 22
2-4 タンパク結合 ... 22
2-5 代謝 ... 23
2-6 尿中排泄 ... 24
2-7 CdGとETVの併用投与時の体内動態 ... 24
第3節 考察 ... 26
第4節 小括 ... 28
第4章 4′-C-cyano-2′-deoxguanosine(CdG)の肝疾患モデルラットにおける
体内動態解析 ... 29
第1節 序 ... 29
第2節 結果 ... 29
2-1 Con A誘発VLIモデルラットの作製 ... 29
2-2 血漿中濃度推移 ... 30
2-3 胃の内容物 ... 32
2-4 臓器分布 ... 33
第3節 考察 ... 34
第4節 小括 ... 36
第5章 4′-C-cyano-2′-deoxguanosine(CdG)の腎疾患モデルラットにおける 体内動態解析 ... 37
第1節 序 ... 37
第2節 結果 ... 37
2-1 CKDモデルラットにおけるCdGの静脈内投与時の体内動態 ... 37
2-2 CKDモデルラットにおけるCdGの経口投与時の体内動態 ... 39
2-2-1 血漿中濃度推移 ... 39
2-2-2 臓器分布 ... 40
2-2-3 尿中排泄 ... 41
第3節 考察 ... 42
第4節 小括 ... 44
第6章 総括 ... 45
実験の部 ... 48
謝辞 ... 57
参考文献 ... 58
第1章 緒論
B型肝炎ウイルス(hepatitis B virus; HBV)は1965年にBlumbergらによりオー ストラリア抗原(後の HBs 抗原)として発見され,その後肝炎の原因ウイルスで あることが報告された 1).HBV について,約半世紀にわたり基礎的・臨床的研究 が進められ,現在では,HB ワクチン接種によりHBVの母子感染の予防に成功し,
HBVに慢性的に感染している 5 歳未満の子供の数は激減した 2,3).しかしながら,
HBV の感染力は極めて強く,世界には未だ約 3 憶人の HBV 持続感染者が存在す ると推定され,年間約 100 万人もの人が HBV に起因した病気で死亡している 4). 日本でも約 150 万人の HBV 持続感染者が存在すると推定されており,その 10~
15 %がB型慢性肝炎を発症し,肝硬変や肝細胞癌へ進行し死亡リスクが高くなる
5–7).従って HBV感染症は,現在においても世界的に重要な健康問題の 1つに位置 づけられている8).
現在,日本では B 型慢性肝炎に対する抗ウイルス治療薬として,インターフェ ロン(IFN)製剤と核酸アナログ製剤が使用されている.IFN 療法は抗ウイルス蛋 白の誘導及び免疫賦活作用により,期間限定的に投与することで持続的効果が得 られる治療である.IFNのメリットとして,核酸アナログ製剤が一般的に長期投与 されるのに対して治療期間が24~48週間と限定されていること,薬剤耐性ウイル スを生じないこと,催奇形性がないこと,セロコンバージョン後(HBe抗原陰性,
HBe抗体陽性の状態)は高率で治療効果が持続することなどがあげられる.また,
デメリットとして副作用が高率かつ多様に起こること,投与のための頻繁な通院 が必要になることなどがあげられるが9),ポリエチレングリコール(PEG)化製剤 の登場により,IFN の血中滞留性が向上し,通院間隔が週 1 回となったことに加 え,発熱・関節痛などのインフルエンザ様症状や倦怠感・食欲低下などの副作用は 従来のIFN製剤より軽度になった.
一方,核酸アナログはラミブジン(lamivudine; LAM),アデホビル(adefovir; ADV),
エンテカビル(entecavir; ETV),テノホビル・ジソプロキシルフマル酸塩(tenofovir disoproxil fumarate; TDF)及びテノホビル・アラフェナミド(tenofovir alafenamide;
TAF)の5種類の薬剤が承認されている(Table 1) 10).核酸アナログは HBV増殖 過程における逆転写酵素を特異的に阻害することでHBV複製過程を直接抑制する 逆転写酵素阻害剤(Nucleoside Analogue Reverse Transcriptase Inhibitor; NRTI)であ り,原則として長期継続投与を必要とする治療である.核酸アナログ製剤のメリッ
トとして,1日1回の経口投与であるため治療が簡便であること,副作用が少ない こと,治療反応例の頻度が非常に高率であることなどがあげられるが,デメリット として投与中止による再燃率が高いため原則長期継続投与であること 11),長期投 与による薬剤耐性変異株の出現及び有害事象が発生する懸念がある.
Table 1. Hepatitis B therapeutic drug (nucleoside analog) in Japan 製品名
(一般名) 発売年 作用機序・製剤特性
ゼフィックス
(LAM) 2000年
活性体;ラミブジン5′-三リン酸
HBV DNA ポリメラーゼに対する競合的拮抗作用と DNA
伸長停止作用.
耐性ウイルスが出現しやすい.
ヘプセラ
(ADV) 2004年
活性体;アデホビル二リン酸
HBV DNAポリメラーゼの選択的阻害作用と DNA伸長停
止作用.
バラクルード
(ETV) 2006年
活性体;エンテカビル三リン酸
HBV DNAポリメラーゼによるプライミング阻害,逆転写
阻害及びHBV DNA合成阻害作用.
空腹時投与.
テノゼット
(TDF) 2014年
活性体;テノホビル二リン酸
HBV DNAポリメラーゼに対する競合的阻害作用.
テノホビルのプロドラッグ.
ベムリディ
(TAF) 2017年
活性体;テノホビル二リン酸
HBV DNAポリメラーゼに対する競合的阻害作用.
テノホビルのプロドラッグ.TDFより細胞透過性が高い.
これらの B 型慢性肝炎に対する核酸アナログの開発背景には薬剤耐性変異株の 出現と安全性の克服が密接に関係している.2000 年に日本で初めて承認された LAMは,投与開始後 6~9か月で LAM耐性ウイルスが出現し始め,6年後には耐 性ウイルスの出現率は 70 %にもなる 12,13).そこで LAM 耐性株にも有効性を示す ADVが開発されたが,ADV耐性ウイルスの出現率は 5年目で 29 %であった14).
また, ADV は重要な副作用である腎機能障害と低リン血症の発症が治療継続の
妨げとなった 15).これらの問題を受け,より強力な高い抗ウイルス活性を有し,
LAM 耐性株にも有効性を示す ETV が開発された 16,17).ETV は大きな副作用は報
告されておらず,耐性ウイルスの出現率は 5 年で 1.2 %であった 18,19).ETV に続 き,従来の核酸アナログに抵抗性及び無効の症例に対しても有効性を示すテノホ ビル(TFV)のプロドラッグであるTDFとTAFが承認された.現時点において TDF とTAF の耐性ウイルスは確認されていない 20,21).従って,現在の核酸アナログを 使用する場合の第一選択薬は,薬剤耐性出現のリスクが少ないETV,TDF 及びTAF となっている10).しかしながら,これらの核酸アナログでは,(i) HBVの増殖を抑 制できても HBV自体を体内から完全に排除することはできないこと,(ii) 核酸ア ナログ中止による肝炎の再燃も高率で起こることから,B 型慢性肝炎患者に対す る核酸アナログの長期投与は避けられない.そのため,今後,ETV・TDF・TAFに 対する薬剤耐性変異株が高頻度で出現する可能性は否定できず,さらに強力な抗 ウイルス効果を有するまたは耐性プロファイルが異なるような新規HBV治療薬の 開発が望まれる.
近年,4′位を修飾した核酸アナログがHBV (ヘパドナウイルス)やヒト免疫不 全ウイルス(Human Immunodeficiency Virus; HIV)(レトロウイルス)に高い抗ウイ ルス活性を有することが報告され22),HBVやHIV感染症の治療薬として開発が盛 んに進められている.実際に 4′位にエチニル基を有する 4′-C-ethynyl-2-fluoro-2′- deoxyadenosine(EFdA)は抗HIV薬として現在臨床試験中である 23).また,4′位を シアノ基で修飾した複数の4′-C-cyano-2′-deoxynucleosides(Fig. 1)はin vitroにお いてHBVに対して高い抗ウイルス活性を有することが明らかとなった 24,25).さら に,これらの4′-C-cyano-2′-deoxynucleosidesはin vivoにおいてもヒト肝キメラマウ スを用いた薬理試験において,十分にHBV DNA 量を減少させるだけでなく,ETV 耐性変異株に対しても高い有効性を示すことが報告されている 25).このように4′- C-cyano-2′-deoxynucleosidesはin vitro及びin vivoにおいてHBVに対して高い抗ウ イルス活性を示し,新規HBV治療薬として期待されることより,さらなる構造最 適化を検討して新規 HBV 治療薬となりうる薬物の開発研究がなされている 26,27).
Fig. 1. Chemical structure of 4′-C-cyano-2′-deoxynucleosides
一般的な創薬のプロセスの中で,細胞や動物を用いた薬効薬理試験,薬物動態試 験,安全性試験などの非臨床試験を行い,ヒトにおける有効性と安全性を裏付け る.その中でも,薬物の吸収(absorption)・分布(distribution)・代謝(metabolism)・
排泄(excretion)といったいわゆる ADME を明らかにする薬物動態試験は,薬物
の効果持続時間などの有効性は勿論,副作用及び薬物間相互作用を考える上でも 重要である.例えば,HBVに高い抗ウイルス活性を有する TFVは,薬物動態試験 によりバイオアベイラビリティが低く経口投与製剤 には適していないことが明ら
かとなり 28–30),バイオアベイラビリティを改善した TDF が TFV のプロドラッグ
として開発された.さらにTDFは標的細胞への分布が少ないことにより TFVの血 中濃度を高く維持する必要があった.このことにより TDFは重大な副作用である 腎障害を惹起したが,TAF は標的細胞内に効率的に取り込まれるように製剤工夫 したことにより, TFVの血中濃度を低く抑えることで安全性の向上につながって
いる 31,32).このように薬物の体内動態特性を基盤として最適な製剤化を行うこと
で薬物の臨床使用を実現したケースも多く見受けられる.従って,現段階で 4′-C- cyano-2′-deoxynucleosides の体内動態の基盤情報を構築することは,4′-C-cyano-2′-
deoxynucleosides の新規 HBV 治療薬開発をスムーズに行うためにも有用であると
考えられる.
Anti-HBV activity (IC50=0.045 µM) Anti-HBV activity (IC50=0.0004 µM) Fig. 2. Chemical structure of 4′-C-cyano-2′-deoxyinosine (CdI: left) and 4′-C-cyano-2′- deoxyguanosine (CdG: right)
このような背景の下,本研究では 4′-C-cyano-2′-deoxynucleosides の中から HBV に対して抗ウイルス活性を有するCdIとCdGについての体内動態解析を液体クロ マトグラフ質量分析計(liquid chromatograph-mass spectrometry; LC/MS)を活用し て行った.第 2 章において CdI,第 3 章においてCdG の体内動態特性を健常ラッ トを用いて検討した.第4 章では実際のHBV感染時を想定してウイルス性肝障害
(Viral Liver Injury; VLI)モデルラットにおけるCdGの体内動態解析を行い,健常 時との体内動態特性の比較検討した.第 5 章では核酸アナログにおいて用量調整 が必要とされる腎機能低下時のCdG の体内動態特性を明らかにした.以下に得ら れた知見を詳述する.
第2章 4′-C-cyano-2′-deoxyinosine(CdI)の健常ラットにおける体内動態解析
第1 節 序
CdIは4′位をシアノ基で修飾したイノシンベースの NRTI であり,HepG2 2.2.15
cellsを用いた抗 HBV活性評価ではIC50値が 0.045 µMを示した.
これまでに,HIV に対して抗ウイルス活性を有するイノシンベースのNRTIであ るジダノシン(ddI)は,酸性溶液中において不安定であるためバイオアベイラビ リティが低いことが体内動態試験で明らかとなり,この問題を解決した腸溶性カ プセル製剤として上市されている33).従って,イノシンの核酸アナログである CdI についても体内動態特性の検討を行うことは今後の製剤化に向けて大いに意義が ある.
そこで本章では,まず生体サンプル中の CdI 濃度を定量するために,LC/MSを 用いた測定法を確立した.次に静脈内及び経口投与後の CdI の動態特性について ラットを用いて検討するとともに,CdI のタンパク結合特性とチトクローム P450
(CYP)による代謝性についてin vitro試験によって評価した.加えて,耐性ウイ ルスへの対策として臨床では薬剤の切り替え や他の薬剤との併用投与が行われる ため 34),B型慢性肝炎治療薬の ETV とCdIの併用投与時の血中動態も検討した.
第2 節 結果
2-1. LC/MSによる CdIの測定法の確立
LC/MS を用いて CdI の定量を行うために,水溶液中での検出条件の設定を行っ
た.CdIは約2分(m/z 278.1)に単一のピークとして検出された(Fig. 3A).次に,
体内動態特性の評価を行う前に,生体サンプルの薬物を生体内成分と分離するこ とが必須であることから,LC/MS で測定するためのサンプルの前処理条件の検討 を行った.生体サンプル中の薬物の前処理法に最も簡易的に用いられる方法とし て除タンパク法がある.メタノールまたはアセトニトリルを用いた有機溶媒変性 法によってタンパクを除き,CdI をターゲットに LC/MS で測定したところ,ピー クのブロード化(Fig. 3B)やピークの消失(Fig. 3C)が起こった.従って,有機溶 媒による除タンパク法はCdIの前処理法として不適であった.
そこで,生体サンプルの前処理法として固相抽出(Solid Phase Extraction; SPE) 法を検討した.SPEカラムは塩基性化合物の抽出に最適な Oasis® MCXを用いて,
CdIの抽出条件を検討した結果, Fig. 4 で示すSPE のプロトコールを用いること で,血漿中サンプルからの内因性の干渉ピークもなくCdIを単離することができ,
血漿からの CdIの抽出に成功した(Fig. 5).さらに血漿からの CdI のSPE回収率 は92.4 ± 10.9 %であった.
Fig. 3. LC/MS chromatograms of (A) CdI spiked with water, (B) after a methanol treatment of CdI spiked with plasma, and (C) after an acetonitrile treatment of CdI spiked with plasma.
Fig. 4. SPE protocol.
Fig. 5. LC/MS chromatograms of (A) blank plasma and (B) CdI spiked with plasma after SPE treatment.
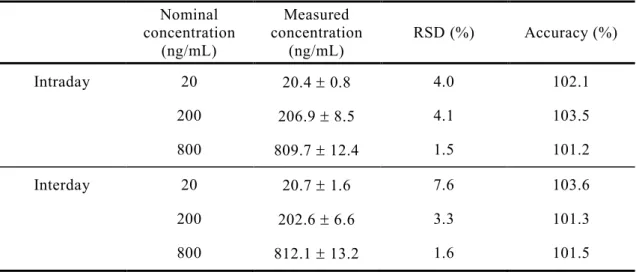
確立したSPE及びLC/MS条件をCdIのin vivo体内動態実験に適用するために,
日内及び日間変動のバリデーションを行った.それぞれの標準濃度(x)に対して CdI(y)のピーク強度をプロットすることにより作成した検量線(6 濃度)は,
15.625-500 ng/mLの濃度範囲で良好な直線性を示し(r2=0.99),定量下限は15.625
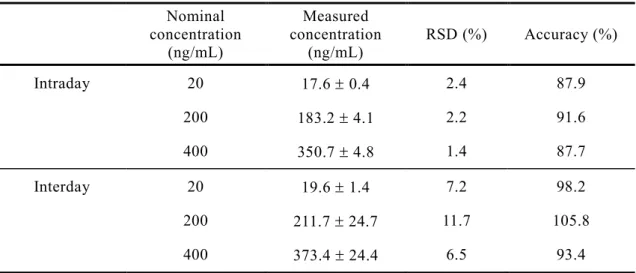
ng/mLだった.日内及び日間の精度と真度をTable 2に示す.日内変動の精度は1.4-
2.4 %,真度は87.7-91.6 %であった.また,日間変動の精度と真度は,それぞれ 6.5-
11.7 %,93.4-105.8 %であり,これらのバリデーションの結果は,SPE法を用いて
確立したLC/MSの測定法の妥当性を示した35).
Table 2. Intra- and inter-day accuracy and precision for CdI in plasma Nominal
concentration (ng/mL)
Measured concentration
(ng/mL) RSD (%) Accuracy (%)
Intraday 20 17.6 0.4 2.4 87.9
200 183.2 4.1 2.2 91.6
400 350.7 4.8 1.4 87.7
Interday 20 19.6 1.4 7.2 98.2
200 211.7 24.7 11.7 105.8
400 373.4 24.4 6.5 93.4
Each value represents the mean ± S.D. (n=3-5)
2-2. 血漿中濃度推移
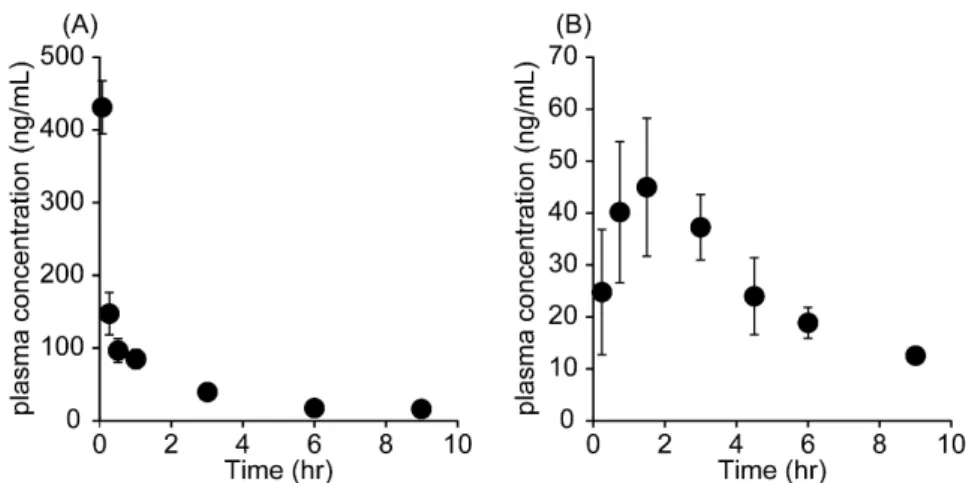
CdIを1 mg/kgの用量でラットに静脈内または経口投与した後の血漿中CdI濃度
をLC/MSを用いて測定した.CdI投与後9時間までの血漿中濃度推移をFig. 6に,
得られた血漿中濃度よりノンコンパートメントモデルを用いて算出した動態学的 パラメーターを Table 3 に示す.CdI の半減期(t1/2)は静脈内投与時に約 3 時間,
経口投与時に約3.6時間であり,経口投与後の最高血中濃度到達時間(tmax)は1.3 時間だった.さらにCdIのバイオアベイラビリティは約 65 %であり,腸管からの 吸収は比較的良好であった.
Fig. 6. Time course for the plasma concentration of CdI after (A) intravenous injection and (B) oral administration at a dose of 1 mg/kg in rats.
Each value represents the mean ± SD. (n=4)
Table 3. Pharmacokinetic parameters of CdI after intravenous and oral administration of a dose of 1 mg/kg in rats.
intravenous oral
t1/2 (hr) 3.01 ± 1.48 3.59 1.20
MRT (hr) 3.58 ± 1.86 5.47 ± 1.52
AUC (hr・ng/mL) 475.4 ± 82.2 307.5 49.7
CL (L/hr/kg) 2.08 ± 0.34 -
CL/F (L/hr/kg) - 3.32 1.52
tmax (hr) - 1.29 0.34
t1/2: half-life, MRT: mean residence time, AUC: area under the concentration-time curve, CL:
clearance, F: bioavailability, tmax: time to reach peak plasma concentration. Each value represents the mean ± S.D. (n=4)
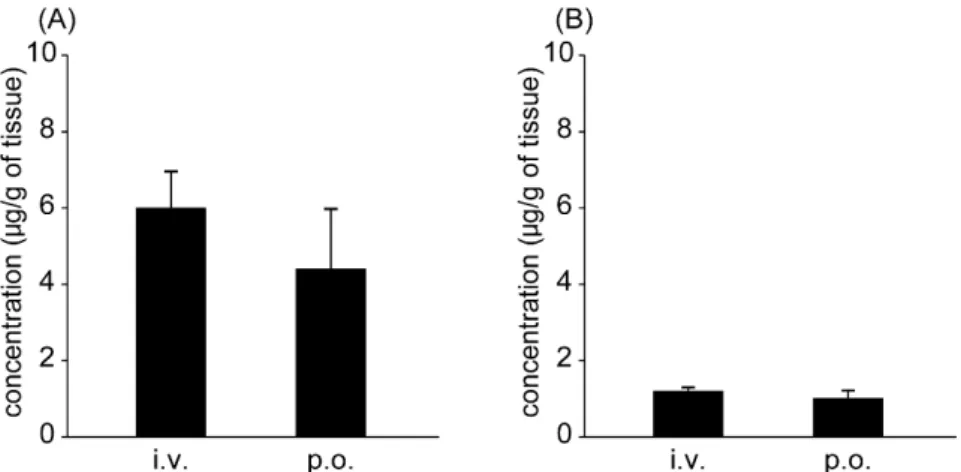
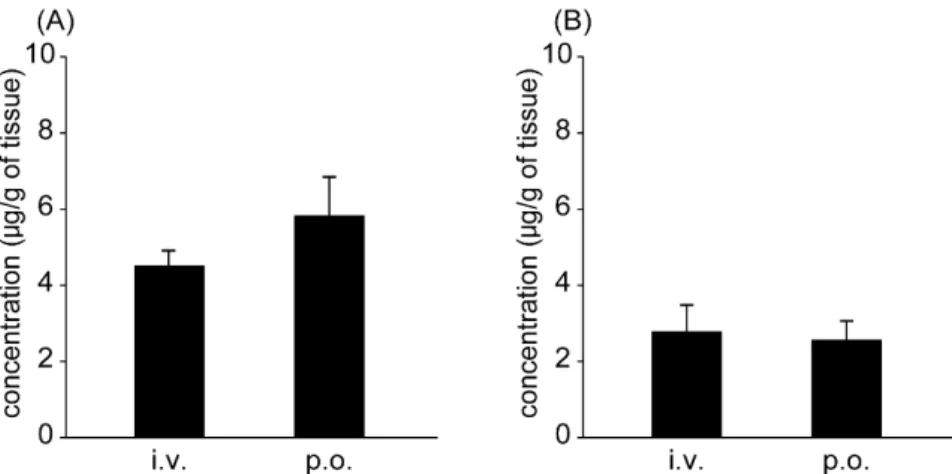
2-3. 臓器分布
CdI投与後,生体内で代謝,排泄を主に担っている肝臓及び腎臓におけるCdIの 分布を評価した.その結果,投与後 9 時間において CdI は腎臓よりも肝臓に多く 分布していた(Fig. 7).
Fig. 7. The tissue distribution to (A) liver and (B) kidney of CdI at 9 hr after intravenous injection (i.v.) and oral administration (p.o.) at a dose of 1 mg/kg in rats.
Each value represents mean ± SD (n=4).
2-4. タンパク結合
血液中で多くの薬物はアルブミンや α1-酸性糖タンパク質(α1-acid glycoprotein;
AGP)に非共有結合する 36).タンパク結合は薬物の効果や薬物間相互作用に深く 関連しており,重要な薬物動態情報の1つである.そこで,CdIのタンパク結合率 を調べるため,限外ろ過法によりヒト血漿中の CdI のタンパク結合率を算出した
ところ 63.7 ± 0.04 %であった.また,薬物のタンパク結合に関連する主要な血漿
タンパク質であるアルブミンとAGPに対するCdIのタンパク結合率を検討した結 果,ヒト血清アルブミン(Human serum albumin; HSA)が14.7 ± 0.03 %,AGPが 22.1 ± 0.01 %であった(Fig. 8).
Fig. 8. The binding rate of CdI to human plasma, HSA and AGP.
Each value represents the mean ± SD (n=3).
2-5. 代謝
CdI が多くの薬物の代謝に関わっている CYP の基質であるか否かを調べるため
に,in vitro においてラット及びヒト肝ミクロソームを用いて代謝アッセイ試験を
NADPH regenerating system を用いて行った.その結果,肝ミクロソーム中でイン
キュベートしたCdIの残存率はラット肝ミクロソーム中で 73.7 ± 10.7 %,ヒト肝 ミクロソーム中で75.1 ± 12.8 %であった(Fig. 9).
Fig. 9. The residual rate of CdI after incubation of CdI in microsomes with NADPH system.
Each value represents the mean ± SD (n=3).
2-6. 尿中排泄
次に,尿中の CdI濃度を測定した.尿中においても CdI(m/z 278.1)の単一のピ ークが検出され,CdI は未変化体として尿中に排泄されることが明らかとなった
(Fig. 10).尿中に排泄されたCdIの未変化体量は,経口投与後 9時間において投
与量の約20 %であった.これは CdIのバイオアベイラビリティ(約 65 %)を考慮 すると,吸収された約 30 %が尿中に排泄された.
Fig. 10. (A) LC/MS chromatograms of CdI in urine. (B) Accumulative amount of CdI excretion in urine until 9 hr after intravenous injection (i.v.) and oral administration (p.o.) at a dose of 1 mg/kg in rats.
Each value represents the mean ± SD (n=4).
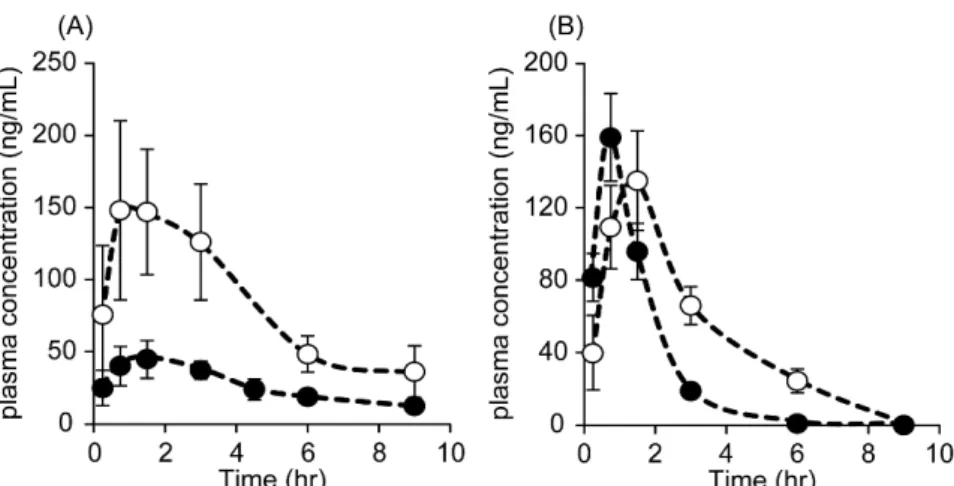
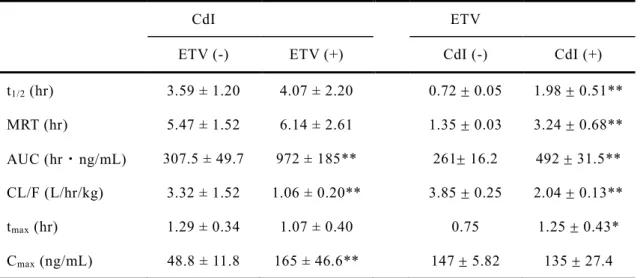
2-7. CdIとETVの併用投与時の体内動態
CdI と ETV の併用投与が ETV 及び CdI の動態特性に及ぼす影響を評価した.
ETV と同時に投与した際の CdI の最高血中濃度(Cmax)及び血中濃度-時間曲線 下面積(AUC)は,CdI単独投与時と比較して大幅に増加した.t1/2及びtmaxはETV との併用投与によってほとんど変化しなかった(Fig. 11A, Table 4).Fig. 11BにETV
(1 mg/kg)単独経口投与後及び CdI(1 mg/kg)を同時経口投与した際の ETV(1
mg/kg)の血漿中濃度推移曲線を示す.CdIを併用投与すると,ETV単独投与時と
比較して ETVのt1/2及び AUCが有意に増加した(Table 4).さらに CdIと併用投 与した場合,ETVの tmaxは遅延した.CdI とETV の併用投与はどちらの薬物もク リアランスの低下とAUCが増大し,単独投与時より血中に長く滞留することが分 かった.
Fig. 11. (A) Time course for the plasma concentration of CdI after the oral administration of CdI alone (closed circle) and combination with ETV (open circle) at doses of 1 mg/kg in rats. (B) Time course for the plasma concentration of ETV after the oral administration of ETV alone (closed circle) and combination with CdI (open circle) at doses of 1 mg/kg in rats.
Each value represents the mean ± SD. (n=4)
Table 4. Pharmacokinetic parameters of CdI and ETV after the oral co-administration of CdI and ETV at a dose of 1 mg/kg in rats.
CdI ETV
ETV (-) ETV (+) CdI (-) CdI (+)
t1/2 (hr) 3.59 ± 1.20 4.07 ± 2.20 0.72 0.05 1.98 0.51**
MRT (hr) 5.47 ± 1.52 6.14 ± 2.61 1.35 0.03 3.24 0.68**
AUC (hr・ng/mL) 307.5 ± 49.7 972 ± 185** 261 16.2 492 31.5**
CL/F (L/hr/kg) 3.32 ± 1.52 1.06 ± 0.20** 3.85 0.25 2.04 0.13**
tmax (hr) 1.29 ± 0.34 1.07 ± 0.40 0.75 1.25 0.43*
Cmax (ng/mL) 48.8 ± 11.8 165 ± 46.6** 147 5.82 135 27.4
*p < 0.05, **p < 0.01 vs. oral administration of CdI or ETV alone, respectively. Each value represents the mean ± SD (n=4).
第3 節 考察
薬物動態試験には放射性同位元素(Radioisotope; RI)を用いる RI法とRIを使用 しない非RI法がある.RI法は検出感度及び定量性が高く,代謝物も含めた薬物の 動きを追跡することが容易であるが,人体や環境への被ばく に配慮する必要があ る.非RI法では,薬物と生体内成分を分離することが必須であり,定量性は RI法 には劣るが,安全かつ簡便に体内動態実験を行うことができる.近年の機器分析で はガスクロマトグラフィー(Gas Chromatography; GC), 高速液体クロマトグラフ ィー(High Performance Liquid Chromatography; HPLC),GC/MS,LC/MSやLC/MS/MS などが薬物の定量に頻繁に用いられており,核酸アナログも例外ではない 37).そ こで,本研究では安全かつ簡便に体内動態実験を行うことを念頭に,本実験に用い る低分子核酸アナログの定量にはLC/MSを使用して,体内動態解析を行った.
まず,生体サンプル中から CdI を抽出するための前処理法を検討した.体内動 態試験において多数のサンプルの処理が必要になることを考慮すると,前処理の 工程は効率よく簡便なものが望ましい.そこで,最も簡易的な前処理法として有機 溶媒を用いる除タンパク法を試みたが,メタノール及びアセトニトリルによる除 タンパクはどちらもCdIの前処理には適していなかった(Fig. 3).除タンパク法は 生体成分のタンパクのみを変性・凝集させることでタンパク質を除去する方法で あることから,イオンや低分子物質などのタンパク質以外の夾雑物は残存する.従 って,CdI のピークのブロード化やピークの消失(Fig. 3)の原因として,生体サ ンプル中の夾雑成分が CdI のイオン化を阻害している可能性が考えられる.そこ で,イノシンベースの NRTIであるddIのLC/MS分析の前処理にSPE法を用いて いたことから 38),前処理法として Oasis® MCX を用いた SPE法の検討を行った.
Oasis® MCXは陽イオン交換-逆相ミックスモード充填剤で,塩基性化合物の抽出
に適しているカラムである.Oasis® MCX を用いた SPE 法により CdI は血漿から
92.4±10.9 %の割合で回収され,生体内成分の干渉もなく単一の CdIのピークを得
ることができた(Fig. 5).また,血漿サンプルにおけるこの定量法の日内及び日間 変動は,生物分析法のガイドライン35)基準である精度15 %以内,真度85~115 % を満たしており(Table 2),本研究で確立した LC/MS 測定条件および前処理法は 生体サンプル中のCdI定量法として検出限界は15.625 ng/mLであり十分であった.
CdIを1 mg/kgの用量で経口投与し得られた血漿中濃度推移曲線からノンコンパ
ートメント解析を使用して,体内動態パラメーターを算出した(Table 3).経口投
与後の CdI の t1/2は約 3.6 時間であり,tmaxは 1.3 時間だった.以前の研究におい て,CdIと同様のイノシンベースの ddIは回腸や結腸よりも十二指腸で吸収される ことが報告されており,部位依存的な吸収を示す 39).加えて,他のプリン構造ベ ースであるETVも十二指腸>空腸>回腸の順で吸収される割合が高いと報告されて いる 40).従って,CdIも ddI やETV のように十二指腸で速やかに吸収されたため 早い tmaxを示したと推測される.CdI のバイオアベイラビリティは約 65 %と ddI
(ラットにおいて 8-16 %)と比較するとはるかに高く 41,42),腸管から比較的効率 的に吸収されることが示され,CdIは経口製剤として使用できると考えられる.
薬物の分布という観点において,タンパク結合特性は薬効や薬物間相互作用に 関連することがあるため,重要な体内動態情報である.本研究で,ヒト血漿中にお けるCdIのタンパク結合率は63.7 ± 0.04 %と低値を示し,タンパク結合は CdIの 体内動態に影響を与えないと考えられる.また,多くの薬物はアルブミンや AGP 非共有結合するが,CdI はアルブミンや AGP との結合率も低かったことから CdI はこれらのタンパク質ではなく他の血漿タンパク質と結合すると考えられた(Fig.
8).さらに,CdI は腎臓より肝臓に多く分布しており,HBV 治療薬として好まし い臓器分布特性を有していた.
肝臓中には多くの代謝酵素が発現しており,特に CYP は多くの薬物の第一相代 謝反応に関与している.そこで,CdIが肝 CYPによって代謝されるかをin vitroに おいて検討した.ラット及びヒト肝ミクロソームを用いて行った代謝実験の結果,
約75 %が残存していたためCdIの代謝にCYPの関与は少ないことが示された(Fig.
9).この結果は,他の核酸アナログが CYPの基質ではないことと合致した 43).さ らに,イノシンはげっ歯類ではアラントインに,ヒトでは尿酸に代謝されることが 知られており 44),イノシン構造を有する CdI も同様の代謝経路を辿る可能性があ る.実際に,イノシン構造を有するddIの代謝経路は既知のプリン代謝経路と同様 であり,最終代謝産物としてアラントイン及び尿酸,ヒポキサンチン,キサンチン などが検出されると報告されている45).従って,CdIの代謝経路は内因性のイノシ ンの代謝経路と類似していると考えられる.
CdIの排泄性について検討するために,尿中のCdI未変化体量を測定した.その 結果,経口投与後9時間までに投与量の約 20 %が尿中に未変化体として排泄され
た(Fig. 10).これに CdIのバイオアベイラビリティ(約 65 %)を加味すると,吸
収されたCdIの約30 %が尿中に未変化体として排泄されたことになる.また,経 口投与 72 時間後までの CdI の尿中への累積排泄量は投与量の約 45 %まで徐々に
増加した.核酸アナログは細胞内で速やかにリン酸化され,生じたリン酸化体はそ の後比較的ゆっくりと脱リン酸化され体循環に戻されることが知られている46,47). そのため,CdIも同様にリン酸化された後に,脱リン酸化され尿中に未変化体とし て排泄されていると考えられる.加えて,先述したように CdI は内因性イノシン と同様にアラントイン様代謝物に代謝されると考えられることから,一部の CdI はアラントイン様代謝物として尿中に排泄されていると考えられる.
B 型慢性肝炎の治療において薬剤耐性変異株が出現し,一定の治療効果が得ら れなくなった場合,他の核酸アナログへの切り替えや他の核酸アナログ を併用投 与する治療を行う34).実際に,プリン構造を有する TFVとddIを同時に投与する と ddI の動態変化を引き起こすことが以前の研究で報告されている 48,49).これは 類似したプリン構造を有したCdIとETVの併用投与が互いの体内動態特性に影響 を与える可能性を示唆する.従って,薬物動態学の観点で CdIが従来の B 型慢性 肝炎に対する核酸アナログ製剤と併用投与できるか検討することは重要である.
そこでCdIとETVを同時に投与した際のそれぞれの体内動態特性への影響を検討 した.その結果,併用投与はどちらの薬物も単独投与した際の体内動態とは異な り,同時併用投与するとCdIとETVのそれぞれの動態特性に影響を与えることが 分かった(Fig. 11).薬物動態学的相互作用が起きた明確な要因は不明であるが,
CdI と ETV の構造が類似していることで同様の体内動態をとることに起因する可 能性は高いと考えられる.先述のように,CdI と ETV はいずれも上部小腸で吸収 されると考えられており40),ETV がトランスポーターを介してCdIの吸収を促進 する可能性がある.さらに,腎臓における核酸アナログの分泌と再吸収には複数の 薬物トランスポーターが関与しており 50,51),排泄経路における競合による消失の 遅延も要因として考えられる.
以上,健常ラットを用いた体内動態解析の結果,CdIは比較的良好な吸収性と肝 臓への分布性を持ち,経口投与に適していることが示された.ただし,類似の構造 をもつ薬物との併用投与は互いの薬物の体内動態特性に影響を及ぼす可能性があ るため,注意が必要である.また,in vivo及びin vitro実験において得られた結果 より代謝及び排泄経路は類似構造を有する既存の薬物と同様であると考えられる.
第4 節 小括
本章では,LC/MS による CdI の定量法を確立し,in vivo 及び in vitro において CdIの体内動態特性を検討した.
以下に得られた知見を要約する.
1. LC/MSを用いたCdIの測定条件を確立した.また,Oasis® MCXを用いたSPE
法により生体サンプルから CdIを高回収率で抽出する前処理法を確立した.
2. 健常ラットにおける体内動態解析の結果,血中濃度推移より CdIの血中滞留性 及びバイオアベイラビリティは比較的良好であることが示された.
3. 血中において CdIはアルブミンや AGPとほとんど結合せず,タンパク結合は CdIの分布特性に影響を与えないことが示された.また,吸収された CdIは腎 臓よりも肝臓に多く分布し,肝 CYP による代謝はほとんど受けないことが示 された.
4. CdI投与9時間後の尿への累積未変化体排泄量は投与量の約 20 %であったが,
72時間後には約 45 %に達しており,徐々に尿中に未変化体として排泄される ことが明らかとなった.
5. CdI と ETV の併用投与ではそれぞれの動態特性に影響を及ぼすことが明らか となった.
以上の結果より,CdIは経口 HBV 治療薬に適した体内動態特性を有しているこ とが分かった.また,CdI は ETV などの構造類似体を同時に投与すると,互いの 体内動態特性に影響を及ぼすことから併用投与には投与時間を変えるなどの注意 が必要であることが示された.