Advanced Laser and Photon Science レーザー・光量子科学特論
First principles simulations 第一原理計算
Takeshi Sato
http://ishiken.free.fr/english/lecture.html [email protected]
Interac(on of atoms and molecules with intense electric fields
(1013 〜1015 W/cm2)
Nuclear aBrac(on & electric field
electron Escape poten(al
barrier
Z
r + E · r
7/5 No. 3
PROGRESS ARTICLE
382 nature physics | VOL 3 | JUNE 2007 | www.nature.com/naturephysics
of the optical pulse controls the kinetic energy2, amplitude17 and phase18 of the recollision electron and therefore the attosecond pulse19 that it produces.
In addition to producing attosecond electron and photon pulses, the recollision simultaneously encodes all information on the electron interference. Once the amplitude and phase of the electron interference is encoded in light, powerful optical methods become available to ‘electron interferometry’.
Classical trajectory calculations show that fi ltering a limited band of photon energies near their maximum (cut-off ) confi nes emission to a fraction of a femtosecond17. Such a burst emerges at each recollision of suffi cient energy. Th e result is a train of attosecond bursts of extreme ultraviolet (XUV) light spaced by Tosc/2 (ref. 1).
For many applications, single attosecond pulses (one burst per laser pulse) are preferred. Th ey emerge naturally from atoms driven by a cosine-shaped laser fi eld comprising merely a few oscillation cycles (few-cycle pulse)3. Th en only the electron pulled back by the central half-wave to its parent ion possesses enough energy to contribute to the fi ltered high-energy emission (Fig. 3). Turning the cosine waveform of the driving laser fi eld into a sinusoidally shaped one (by simply shift ing the carrier wave with respect to the pulse peak8) changes attosecond photon emission markedly: instead of a single pulse, two identical bursts are transmitted through the XUV bandpass fi lter. Controlling the waveform of light8 has proved critical for controlling electronic motion and photon emission on an attosecond timescale and permitting the reproducible generation of single attosecond pulses19.
Th e shortest duration of a single attosecond pulse is limited by the bandwidth within which only the most energetic recollision contributes to the emission. In a 5-fs, 750-nm laser pulse this bandwidth relative the emitted energy is about 10%. At photon energies of ~100 eV this translates into a bandwidth of ~10 eV, allowing pulses of about 250 attoseconds in duration17. At a photon energy of 1 keV (ref. 20) a driver laser fi eld with the above properties will lead to single pulse emission over roughly a 100-eV band, which
may push the frontiers of attosecond technology near the atomic unit of time, 24 attoseconds. Manipulating the polarization state of the driver pulse17 enables the relative bandwidth of single pulse emission to be broadened21,22 by ‘switching off ’ recollision before and aft er the main event. Together with dispersion control23, this technique has recently resulted in near-single-cycle 130-attosecond pulses at photon energies below 40 eV (ref. 24). Confi ning tunnel ionization to a single wave crest at the pulse peak constitutes yet another route to restricting the number of recollisions to one per laser pulse. Superposition of a strong few-cycle near-infrared laser pulse with its (weaker) second harmonic25,26 is a simple and eff ective way of achieving this goal.
Th is attosecond-pulsed XUV radiation emerges coherently from a large number of atomic dipole emitters. Th e coherence is the result of the atomic dipoles being driven by a (spatially) coherent laser fi eld and the coherent nature of the electronic response of the ionizing atoms discussed above. Th e pulses are highly collimated, laser-like beams, emitted collinearly with the driving laser pulse. Th e next section addresses the concepts that allowed full characterization of the attosecond pulses.
MEASUREMENT TECHNOLOGY
Any pulse measurement method must directly or indirectly compare the phase of diff erent Fourier components of a pulse.
Autocorrelation, SPIDER and FROG, three extensively used methods to characterize optical pulses27, use nonlinear optics to shift the frequency of the Fourier components diff erentially so that neighbouring frequency components can be compared.
Th e electron-optical streak camera — an older ultrafast pulse
Pulse duration (fs)
1970 1980
10–1 100 101 102 103 104 105
1990 Year
2000 2010
Figure 1 Shorter and shorter. The minimum duration of laser pulses fell continually from the discovery of mode-locking in 1964 until 1986 when 6-fs pulses
were generated. Each advance in technology opened new fi elds of science for
measurement. Each advance in science strengthened the motivation for making even shorter laser pulses. However, at 6 fs (three periods of light), a radically different
technology was needed. Its development took 15 years. Now attosecond technology is providing radically new tools for science and is yet again opening new fi elds for real-time measurement. Reprinted in part, with permission from ref. 65.
Ψg
Ψc = a(k)eikx–iωt
30 Å
Figure 2 Creating an attosecond pulse. a–d, An intense femtosecond near-infrared or visible (henceforth: optical) pulse (shown in yellow) extracts an electron wavepacket from an atom or molecule. For ionization in such a strong fi eld (a), Newton’s
equations of motion give a relatively good description of the response of the electron.
Initially, the electron is pulled away from the atom (a, b), but after the fi eld reverses, the electron is driven back (c) where it can ‘recollide’ during a small fraction of the laser oscillation cycle (d). The parent ion sees an attosecond electron pulse. This electron can be used directly, or its kinetic energy, amplitude and phase can be
converted to an optical pulse on recollision12. e, The quantum mechanical perspective.
Ionization splits the wavefunction: one portion remains in the original orbital, the other portion becomes a wave packet moving in the continuum. The laser fi eld moves the wavepacket much as described in a–d, but when it returns the two portions of the wavefunction overlap. The resulting dynamic interference pattern transfers the kinetic energy, amplitude and phase from the recollision electron to the photon.
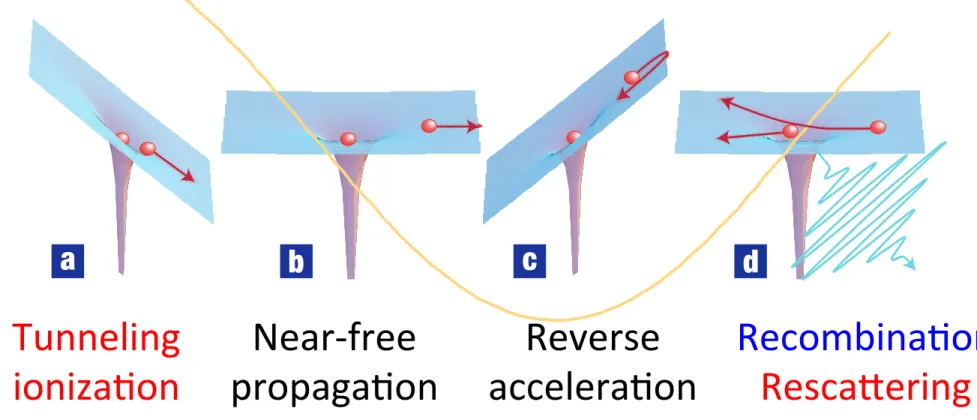
Femtosecond intense laser fields (Visible〜IR, 1013 〜1015 W/cm2)
Tunneling
ioniza(on Near-free
propaga(on Reverse
accelera(on Recombina(on RescaBering
Corkum, Phys. Rev. LeB. 71, 1994 (1993)
7/5 No. 4
PROGRESS ARTICLE
382 nature physics | VOL 3 | JUNE 2007 | www.nature.com/naturephysics
of the optical pulse controls the kinetic energy2, amplitude17 and phase18 of the recollision electron and therefore the attosecond pulse19 that it produces.
In addition to producing attosecond electron and photon pulses, the recollision simultaneously encodes all information on the electron interference. Once the amplitude and phase of the electron interference is encoded in light, powerful optical methods become available to ‘electron interferometry’.
Classical trajectory calculations show that fi ltering a limited band of photon energies near their maximum (cut-off ) confi nes emission to a fraction of a femtosecond17. Such a burst emerges at each recollision of suffi cient energy. Th e result is a train of attosecond bursts of extreme ultraviolet (XUV) light spaced by Tosc/2 (ref. 1).
For many applications, single attosecond pulses (one burst per laser pulse) are preferred. Th ey emerge naturally from atoms driven by a cosine-shaped laser fi eld comprising merely a few oscillation cycles (few-cycle pulse)3. Th en only the electron pulled back by the central half-wave to its parent ion possesses enough energy to contribute to the fi ltered high-energy emission (Fig. 3). Turning the cosine waveform of the driving laser fi eld into a sinusoidally shaped one (by simply shift ing the carrier wave with respect to the pulse peak8) changes attosecond photon emission markedly: instead of a single pulse, two identical bursts are transmitted through the XUV bandpass fi lter. Controlling the waveform of light8 has proved critical for controlling electronic motion and photon emission on an attosecond timescale and permitting the reproducible generation of single attosecond pulses19.
Th e shortest duration of a single attosecond pulse is limited by the bandwidth within which only the most energetic recollision contributes to the emission. In a 5-fs, 750-nm laser pulse this bandwidth relative the emitted energy is about 10%. At photon energies of ~100 eV this translates into a bandwidth of ~10 eV, allowing pulses of about 250 attoseconds in duration17. At a photon energy of 1 keV (ref. 20) a driver laser fi eld with the above properties will lead to single pulse emission over roughly a 100-eV band, which
may push the frontiers of attosecond technology near the atomic unit of time, 24 attoseconds. Manipulating the polarization state of the driver pulse17 enables the relative bandwidth of single pulse emission to be broadened21,22 by ‘switching off ’ recollision before and aft er the main event. Together with dispersion control23, this technique has recently resulted in near-single-cycle 130-attosecond pulses at photon energies below 40 eV (ref. 24). Confi ning tunnel ionization to a single wave crest at the pulse peak constitutes yet another route to restricting the number of recollisions to one per laser pulse. Superposition of a strong few-cycle near-infrared laser pulse with its (weaker) second harmonic25,26 is a simple and eff ective way of achieving this goal.
Th is attosecond-pulsed XUV radiation emerges coherently from a large number of atomic dipole emitters. Th e coherence is the result of the atomic dipoles being driven by a (spatially) coherent laser fi eld and the coherent nature of the electronic response of the ionizing atoms discussed above. Th e pulses are highly collimated, laser-like beams, emitted collinearly with the driving laser pulse. Th e next section addresses the concepts that allowed full characterization of the attosecond pulses.
MEASUREMENT TECHNOLOGY
Any pulse measurement method must directly or indirectly compare the phase of diff erent Fourier components of a pulse.
Autocorrelation, SPIDER and FROG, three extensively used methods to characterize optical pulses27, use nonlinear optics to shift the frequency of the Fourier components diff erentially so that neighbouring frequency components can be compared.
Th e electron-optical streak camera — an older ultrafast pulse
Pulse duration (fs)
1970 1980
10–1 100 101 102 103 104 105
1990 Year
2000 2010
Figure 1 Shorter and shorter. The minimum duration of laser pulses fell continually from the discovery of mode-locking in 1964 until 1986 when 6-fs pulses
were generated. Each advance in technology opened new fi elds of science for
measurement. Each advance in science strengthened the motivation for making even shorter laser pulses. However, at 6 fs (three periods of light), a radically different
technology was needed. Its development took 15 years. Now attosecond technology is providing radically new tools for science and is yet again opening new fi elds for real-time measurement. Reprinted in part, with permission from ref. 65.
Ψg
Ψc = a(k)eikx–iωt
30 Å
Figure 2 Creating an attosecond pulse. a–d, An intense femtosecond near-infrared or visible (henceforth: optical) pulse (shown in yellow) extracts an electron wavepacket from an atom or molecule. For ionization in such a strong fi eld (a), Newton’s
equations of motion give a relatively good description of the response of the electron.
Initially, the electron is pulled away from the atom (a, b), but after the fi eld reverses, the electron is driven back (c) where it can ‘recollide’ during a small fraction of the laser oscillation cycle (d). The parent ion sees an attosecond electron pulse. This electron can be used directly, or its kinetic energy, amplitude and phase can be
converted to an optical pulse on recollision12. e, The quantum mechanical perspective.
Ionization splits the wavefunction: one portion remains in the original orbital, the other portion becomes a wave packet moving in the continuum. The laser fi eld moves the wavepacket much as described in a–d, but when it returns the two portions of the wavefunction overlap. The resulting dynamic interference pattern transfers the kinetic energy, amplitude and phase from the recollision electron to the photon.
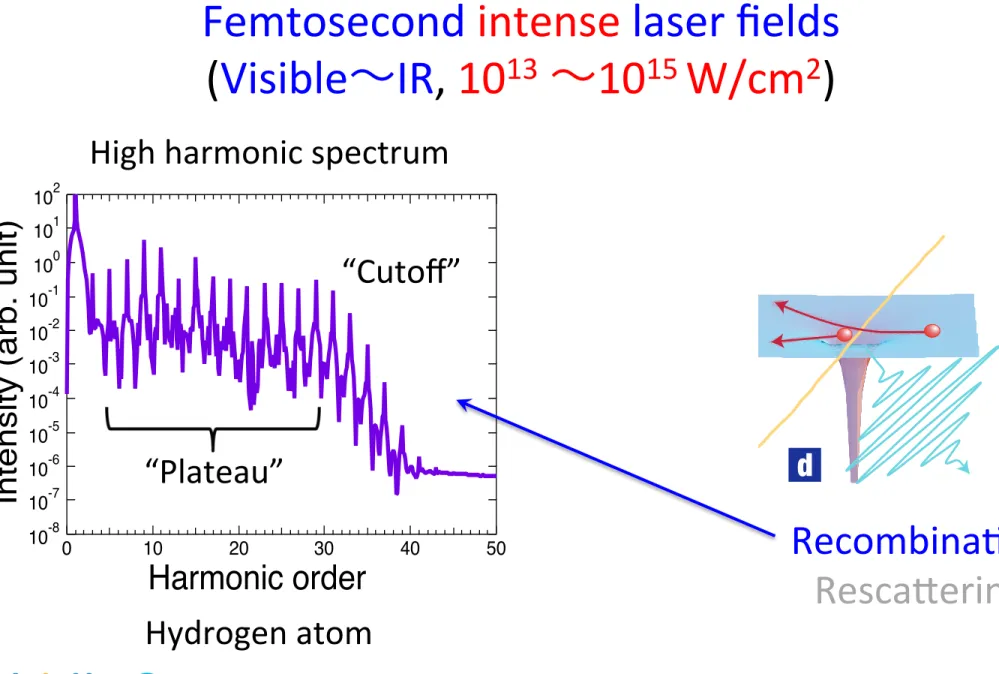
10-8 10-7 10-6 10-5 10-4 10-3 10-2 10-1 100 101 102
Intensity (arb. unit)
50 40
30 20
10 0
Harmonic order
High harmonic spectrum
“Plateau”
“Cutoff”
Hydrogen atom
Femtosecond intense laser fields (Visible〜IR, 1013 〜1015 W/cm2)
Recombina(on RescaBering
4
10-8 10-7 10-6 10-5 10-4 10-3 10-2 10-1 100 101 102
Intensity (arb. unit)
50 40
30 20
10 0
Harmonic order
High harmonic spectrum
“Plateau”
Ioniza(on Poten(al (IP) Kine(c energy
@ return (me (Kr)
Cutoff energy
~! = IP + Kr IP + 3.17Up Ponderomo(ve energy
Up = E02 4!02 :
Recombina(on
E(t) = E0 cos(!0t) Laser field:
“Cutoff”
Hydrogen atom
Femtosecond intense laser fields (Visible〜IR, 1013 〜1015 W/cm2)
7/5 No. 6
PROGRESS ARTICLE
382 nature physics | VOL 3 | JUNE 2007 | www.nature.com/naturephysics
of the optical pulse controls the kinetic energy2, amplitude17 and phase18 of the recollision electron and therefore the attosecond pulse19 that it produces.
In addition to producing attosecond electron and photon pulses, the recollision simultaneously encodes all information on the electron interference. Once the amplitude and phase of the electron interference is encoded in light, powerful optical methods become available to ‘electron interferometry’.
Classical trajectory calculations show that fi ltering a limited band of photon energies near their maximum (cut-off ) confi nes emission to a fraction of a femtosecond17. Such a burst emerges at each recollision of suffi cient energy. Th e result is a train of attosecond bursts of extreme ultraviolet (XUV) light spaced by Tosc/2 (ref. 1).
For many applications, single attosecond pulses (one burst per laser pulse) are preferred. Th ey emerge naturally from atoms driven by a cosine-shaped laser fi eld comprising merely a few oscillation cycles (few-cycle pulse)3. Th en only the electron pulled back by the central half-wave to its parent ion possesses enough energy to contribute to the fi ltered high-energy emission (Fig. 3). Turning the cosine waveform of the driving laser fi eld into a sinusoidally shaped one (by simply shift ing the carrier wave with respect to the pulse peak8) changes attosecond photon emission markedly: instead of a single pulse, two identical bursts are transmitted through the XUV bandpass fi lter. Controlling the waveform of light8 has proved critical for controlling electronic motion and photon emission on an attosecond timescale and permitting the reproducible generation of single attosecond pulses19.
Th e shortest duration of a single attosecond pulse is limited by the bandwidth within which only the most energetic recollision contributes to the emission. In a 5-fs, 750-nm laser pulse this bandwidth relative the emitted energy is about 10%. At photon energies of ~100 eV this translates into a bandwidth of ~10 eV, allowing pulses of about 250 attoseconds in duration17. At a photon energy of 1 keV (ref. 20) a driver laser fi eld with the above properties will lead to single pulse emission over roughly a 100-eV band, which
may push the frontiers of attosecond technology near the atomic unit of time, 24 attoseconds. Manipulating the polarization state of the driver pulse17 enables the relative bandwidth of single pulse emission to be broadened21,22 by ‘switching off ’ recollision before and aft er the main event. Together with dispersion control23, this technique has recently resulted in near-single-cycle 130-attosecond pulses at photon energies below 40 eV (ref. 24). Confi ning tunnel ionization to a single wave crest at the pulse peak constitutes yet another route to restricting the number of recollisions to one per laser pulse. Superposition of a strong few-cycle near-infrared laser pulse with its (weaker) second harmonic25,26 is a simple and eff ective way of achieving this goal.
Th is attosecond-pulsed XUV radiation emerges coherently from a large number of atomic dipole emitters. Th e coherence is the result of the atomic dipoles being driven by a (spatially) coherent laser fi eld and the coherent nature of the electronic response of the ionizing atoms discussed above. Th e pulses are highly collimated, laser-like beams, emitted collinearly with the driving laser pulse. Th e next section addresses the concepts that allowed full characterization of the attosecond pulses.
MEASUREMENT TECHNOLOGY
Any pulse measurement method must directly or indirectly compare the phase of diff erent Fourier components of a pulse.
Autocorrelation, SPIDER and FROG, three extensively used methods to characterize optical pulses27, use nonlinear optics to shift the frequency of the Fourier components diff erentially so that neighbouring frequency components can be compared.
Th e electron-optical streak camera — an older ultrafast pulse
Pulse duration (fs)
1970 1980
10–1 100 101 102 103 104 105
1990 Year
2000 2010
Figure 1 Shorter and shorter. The minimum duration of laser pulses fell continually from the discovery of mode-locking in 1964 until 1986 when 6-fs pulses
were generated. Each advance in technology opened new fi elds of science for
measurement. Each advance in science strengthened the motivation for making even shorter laser pulses. However, at 6 fs (three periods of light), a radically different
technology was needed. Its development took 15 years. Now attosecond technology is providing radically new tools for science and is yet again opening new fi elds for real-time measurement. Reprinted in part, with permission from ref. 65.
Ψg
Ψc = a(k)eikx–iωt
30 Å
Figure 2 Creating an attosecond pulse. a–d, An intense femtosecond near-infrared or visible (henceforth: optical) pulse (shown in yellow) extracts an electron wavepacket from an atom or molecule. For ionization in such a strong fi eld (a), Newton’s
equations of motion give a relatively good description of the response of the electron.
Initially, the electron is pulled away from the atom (a, b), but after the fi eld reverses, the electron is driven back (c) where it can ‘recollide’ during a small fraction of the laser oscillation cycle (d). The parent ion sees an attosecond electron pulse. This electron can be used directly, or its kinetic energy, amplitude and phase can be
converted to an optical pulse on recollision12. e, The quantum mechanical perspective.
Ionization splits the wavefunction: one portion remains in the original orbital, the other portion becomes a wave packet moving in the continuum. The laser fi eld moves the wavepacket much as described in a–d, but when it returns the two portions of the wavefunction overlap. The resulting dynamic interference pattern transfers the kinetic energy, amplitude and phase from the recollision electron to the photon.
Recombina(on RescaBering
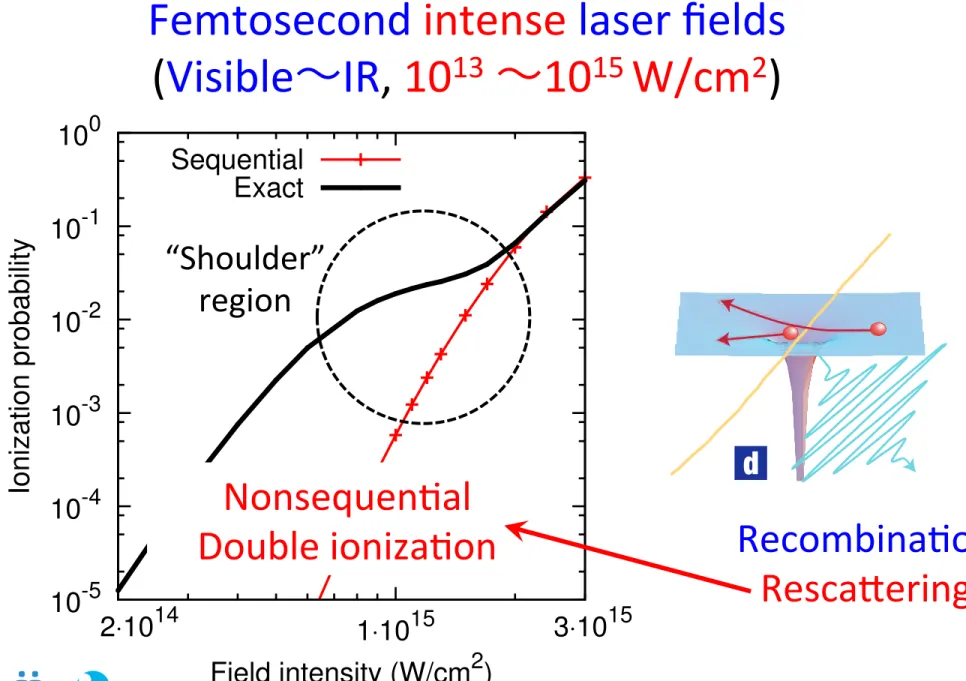
10-5 10-4 10-3 10-2 10-1 100
1⋅1015
Ionization probability
Field intensity (W/cm2)
2⋅1014 3⋅1015
Sequential Exact
Nonsequen(al Double ioniza(on
“Shoulder”
region
Femtosecond intense laser fields (Visible〜IR, 1013 〜1015 W/cm2)
Time-dependent Schrödinger equa(on
First principles simula(ons
H(t) = XN
i=1
( r2i
2 + XM
A=1
ZA
|ri RA| + Vlaser(ri;t) )
+ XN
i=1
X
j<i
1
|ri rj|
i ˙ (r1, r2, . . . rN, t) = H (r1, r2, . . . rN, t)
Computa(onal cost grows factorially w.r.t number of electrons:
Feasible only for H (1 electron), and to a lesser extent He (2 electrons) Time-independent (TID) theories for ground-state: Well established
ü Density-func(onal theory, Coupled-cluster, Many-body Perturba(on theory…
ü Atoms, molecules, solids
Time-dependent (TD) theories for non-sta(onary (excited &
con(nuum) states: Far less developed
Single ac(ve electron (SAE) approxima(on Time-dependent Hartree-Fock (TDHF)
Time-dependent density-func(onal theory (TDDFT)
SAE: CANNOT describe
ü Mul(electron dynamics ü Mul(channel ioniza(on
i ˙(r) = [h(r) + ve↵(r)] (r)
ve↵
TDHF & TDDFT: CANNOT properly describe ü Tunneling ioniza(on
ü Electron correla(on
Mul&configura&on Time-Dependent Hartree-Fock (MCTDHF)
1. Two electron systems (Today)
2. Second quan&za&on (Today/Next week) 3. Mul&configura&on &me-dependent
Hartree-Fock method (Next week)
1. Two electron systems 2. Second quan&za&on
3. Mul&configura&on &me-dependent Hartree-Fock method
What is the simplest-possible wavefunction?
Tunneling ionization
HF(r1, r2) = 1(r1) 1(r2)
i 1(r, t) =
h(r, t) + Z
dr2 | 1(r2, t)|2
|r r2| 1(r, t) TDHF cannot properly describe TI!
At least two orbitals are required
At least two orbitals are required
Tunneling ionization
GVB(r1, r2) / 1(r1) 2(r2) + 2(r1) 1(r2)
GVB(r1, r2) / 1(r1) 2(r2) + 2(r1) 1(r2)
Generalized Valence Bond (Chemistry) Extended Hartree-Fock (Physics)
S12 ⌘ h 1| 2i 6= 0
Tunneling ionization
↵12 = h11|12i ES12
↵21 = h22|21i ES21
E = h12|12i + h12|21i S12h22|21i S21h11|12i det(S)
i
1 S21 S12 1
˙1
˙2 =
h + J2 + K2 S21h
S12h h + J1 + K1
1
2
E ↵21 + ES21
↵21 + ES12 E
1 2
Sato and Ishikawa, J. Phys. B 47 (20), 204031 (2014).
H =
X2
i=1
"
1 2
d2 dx2i
p 2
x2i + 1 + xiE(t) sin!t
#
+ 1
p(x1 x2)2 + 1
0.1 0.2 0.3 0.4 0.5 0.6
ψ∗ψ
GVB orbital 1 GVB orbital 2 HF orbital
+2e
Absorber Absorber
Ground-state
-e -e
-0.06 -0.04 -0.02 0.00 0.02 0.04 0.06 0.08 0.10
amplitude / au
2.6 fs
Field
Model simula(on
0.8 PW/cm2
H =
X2
i=1
"
1 2
d2 dx2i
p 2
x2i + 1 + xiE(t) sin!t
#
+ 1
p(x1 x2)2 + 1
Model simula(on
|h (0)| (t)i|2 h (t)|(x1+x2)| (t)i
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
0 1 2 3 4 5 6 7 8
ground state probability
time / optical cycle TDSEHF
GVB
-20 -10 0 10 20 30
0 1 2 3 4 5 6 7 8
dipole moment / au
time / optical cycle TDSEHF
GVB
H =
X2
i=1
"
1 2
d2 dx2i
p 2
x2i + 1 + xiE(t) sin!t
#
+ 1
p(x1 x2)2 + 1
Model simula(on
GVB(r1, r2) / 1(r1) 2(r2) + 2(r1) 1(r2)
0.00 0.60
-8.0 -4.0 0.0 4.0 8.0
orb. 1
0.00 0.60
-8.0 -4.0 0.0 4.0 8.0
orb. 1 orb. 2
0.00 0.01
-200 -100 0 100 200 t = 1.75T
0.00 0.01
-200 -100 0 100 200 t = 1.75T
0.00 0.01
-200 -100 0 100 200 t = 2.00T
0.00 0.01
-200 -100 0 100 200 t = 2.00T
0.00 0.01
-200 -100 0 100 200 t = 2.25T
0.00 0.01
-200 -100 0 100 200 t = 2.25T
0.00 0.01
-200 -100 0 100 200 t = 2.50T
0.00 0.01
-200 -100 0 100 200 t = 2.50T
0.00 0.01
-200 -100 0 100 200 t = 2.75T
0.00 0.01
-200 -100 0 100 200 t = 2.75T
x (a.u.)
amplitude(a.u.)
TD-GVB TDHF
1(t) 1(t), 2(t)
-20 0 20 40 60
0 1 2 3 4 5 6
-0.2 -0.1 0.0 0.1 0.2
dipole (a.u.) electric field (a.u.)
time (optical cycle) Field
1
Dipole moment (mean posi(on)
TDHF:
electrons 1 & 2
TD-GVB electron 1
TD-GVB electron 2
At least two orbitals required
Tunneling ionization
GVB(r1, r2) / 1(r1) 2(r2) + 2(r1) 1(r2)
GVB(r1, r2) / 1(r1) 2(r2) + 2(r1) 1(r2)
Generalized Valence Bond (Chemistry) Extended Hartree-Fock (Physics)
S12 ⌘ h 1| 2i 6= 0
However, non-orthogonal orbitals result in complicated EOMs
Orbital redundancy
+
A1 + A2
1 2 + 2 1 = C1 1 1 + C3 { 1 2 + 2 1}
GVB(r1,r2) = 1
p2 [ 1(r1) 2(r2) + 2(r1) 1(r2)]
= A1 1(r1) 1(r2) + A2 2(r1) 2(r2)
A1 = 1 + |S12| p2(1 + |S12|2)
S12⇤
|S12|, A2 = 1 |S12|
p2(1 + |S12|2)
S12
|S12|,
1 = 1
p2(1 + |S12|)
⇢ S12
|S12| 1 + 2
2 = 1
p2(1 + |S |)
⇢ S12⇤
|S12| 2 1
Linear transformation of orbitals, that leaves total wavefunction invariant.
Orbital redundancy
close + open
+ B1 + B3 +
A1 + A2 C1 + C2 + C3 +
close + close MCTDHF
Non-orthogonal
1 2 + 2 1 = C1 1 1 + C3 { 1 2 + 2 1}
C1 1 1 + C2 2 2, h 1| 2i = 0 C1 1 1 + C2 2 2, h 1| 2i = 0
C1 1 1 + C2 2 2, h 1| 2i = 0
|h (0)| (t)i|2 h (t)|(x1+x2)| (t)i
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
0 1 2 3 4 5 6 7 8
ground state probability
time / optical cycle TDSEGVB APSG(2)
-20 -10 0 10 20 30
0 1 2 3 4 5 6 7 8
dipole moment / au
time / optical cycle TDSEGVB APSG(2)
(t) = P2
C (t) (1, t) (2, t) / (1, t) (2, t) + (1, t) (2, t)
MCTDHF method for two electron systems
ü Propagate both CI coefficients and orbitals ü Can improve accuracy by increasing orbitals
(t) = P2
ij Cij(t) i(1, t) j(2, t) / 1(1,t) 2(2, t) + 2(1, t) 1(2, t)
= C1 + C2 + C3 + C4
+ …..
+ C5 + C6 (r1, r2)
(r1, r2, t) =
Xn ij
Cij(t) i(r1, t) j(r2, t)
Sequen(al model:
“Shoulder” region:
RescaBering Sequen(al
model:
He à He+ à He2+
PRL, 78, 1884 (1997)
7/5 No. 25
-3 -2 -1 0 1 2 3
10-5 10-4 10-3 10-2 10-1 100
1.0
ionization yield
intensity (1015 W/cm2)
0.2 3.0
Sequential Exact
Double
ioniza(on yields versus intensity
25
Nonsequen(al double ioniza(on (NSDI)
“Shoulder”
region
Nonsequen(al Sequen(al
Strong Electron correla(on
10-5 10-4 10-3 10-2 10-1 100
1.0 × 1015 Intensity (W/cm2) TDSE
He+
HF: N = 1 GVB: N = 2 N = 4 N = 8 N = 16 N = 28
10-5 10-4 10-3 10-2 10-1
100
1.0 × 1015 Intensity (W/cm2) TDSE
HF: N = 1 EHF: N = 2 N = 4 N = 8
He He
(r1,r2,t) =
Xn
Cij(t) i(r1,t) j(r2, t)
Summary
ü At least two non-orthogonal orbitals are required for describing tunneling ionization
GVB(r1, r2) / 1(r1) 2(r2) + 2(r1) 1(r2)
ü Use of orbital redundancy allows reformulation with orthonormal orbitals: MCTDHF method
(r1, r2, t) =
Xn
ij
Cij(t) i(r1, t) j(r2, t)
i. Propagate both CI coefficients and orbitals
ii. Can be generalized to more-than-two electrons
1. Two electron systems (Today)
2. Second quan&za&on (Today/Next week) 3. Mul&configura&on &me-dependent
Hartree-Fock method (Next week)
1 2 3 4 5
Mul(configura(on TDHF (MCTDHF) [Kato and Kono, Caillat et al., …]
= C1 + C2 + C3 + C4 + …..
Superposi(on of ALL Slater determinants within the given number of orbitals
Second quan(za(on
Complete set of one-electron wavefunc(ons (spin-orbitals)
=> Complete set of N-electron Slater determinants
H(t) = XN
i=1
( r2i
2 + XM
A=1
ZA
|ri RA| + Vlaser(ri;t) )
+ XN
i=1
X
j<i
1
|ri rj|
i ˙ (r1, r2, . . . rN, t) = H (r1, r2, . . . rN, t)
= XN
i=1
h(ri, t) + 1 2
XN
i=1
X
j6=i
V (ri,rj)
(r1,r2, ...rN, t) = X
I
CI(t) I(r1,r2, ...rN)
iC˙I(t) = X
I0
h I|H(t)| I0iCI0
Z
Second quan(za(on
Second quan(za(on (r1,r2, ...rN, t) = X
I
CI(t) I(r1,r2, ...rN)
iC˙I(t) = X
I0
h I|H(t)| I0iCI0
h I|H(t)| I0i = Z
⇤I(r1,r2, ...rN)H(r1,r2, ...rN, t) I0(r1,r2, ...rN)dr1dr2 · · · drN
| (t)i = X
n
Cn(t)|ni, iC˙n(t) = X
m
hn|Hˆ (t)|miCm
Hˆ(t) = X
µ⌫
hµ⌫ (t)ˆa†µaˆ⌫ + 1 2
X
µ⌫
V⌫µ aˆ†µaˆ†aˆ ˆa⌫
nµ = {0, 1}
|ni = |n1n2 · · · n1i ⌘ Q1
µ=1(a†µ)nµ|i