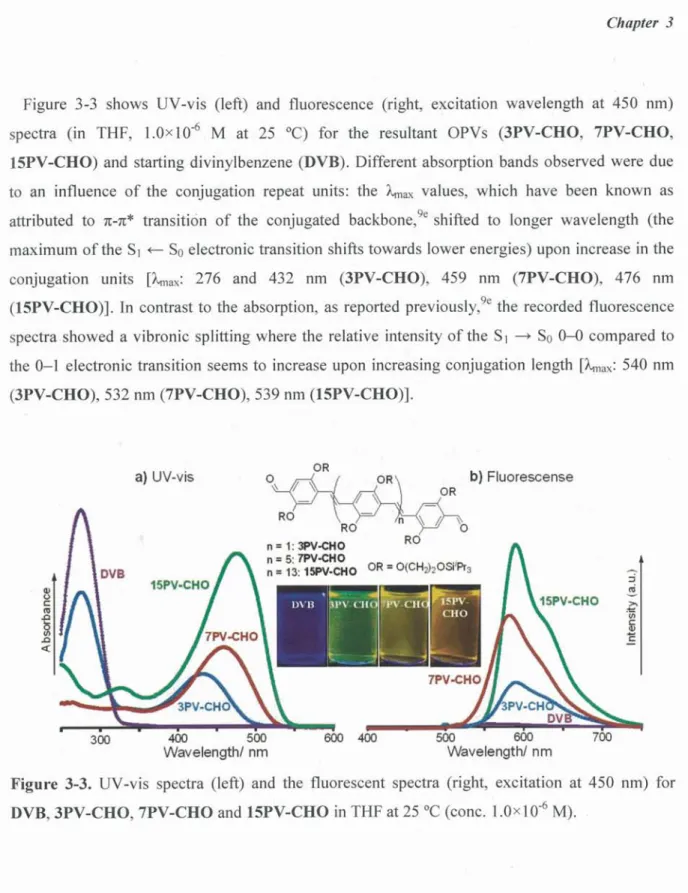
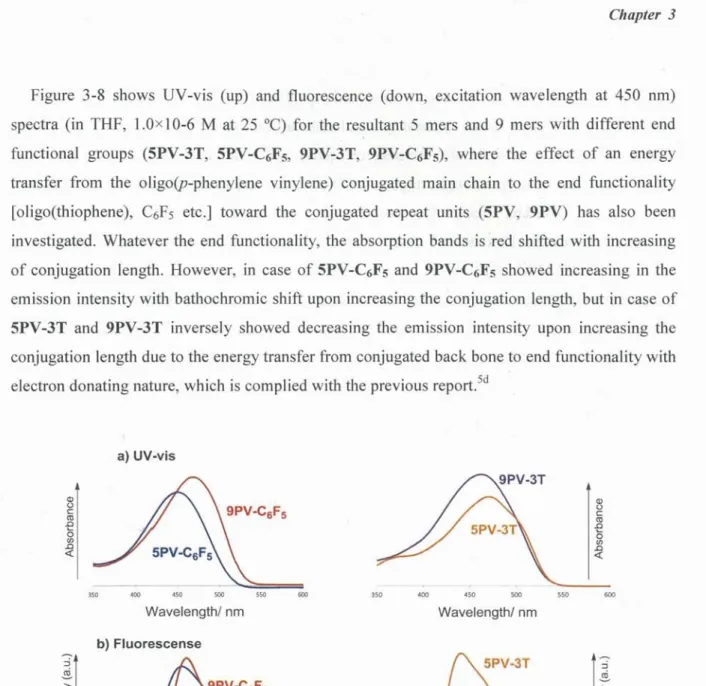
リポ ジ ト リ
※ 年
、 盗 粛/非 公 開
月 日以 降 許 諾 す る
誕
遜
i
,r璽分 子物 質 化 学1480
MohamedMehawedAb dellatif
氏 名 モハメッド メハーウェド アブ
(オ レ フ ィ ン メ タ セ シ ス を利 用 した 末 端 官 能 基 化 共 役 ポ リマ ー/オ リ ゴマ ー の 精 密 合 成(英 文)
Abdellatif,MohamedMehawed
FacultyofScienceandEngineering TokyoMetropolitanUniversity
2013
論 文 名 オ レフ ィン メ タセ シ ス を利 用 した末 端 官 能 基 化 共 役 ポ リ マー/オ リゴマー の精密 合成(英 文)
著 者 モ ハ メ ツ ド メ ハー ウ ェ ド
審 査 担 当者
主 査 動 材%ム
委 員
委 員 吻 巻 喜 委 員 卸 く畝 須
上記 の論 文 を合 格 と判定 す る 平成2夕 年3月2夕 日
首都 大 学東 京大 学 院理 工学研 究科教 授 会
研究科長
1舞 や 望
TOKYO METROPOLITAN UNIVERSITY
TITLE : Precise Synthesis of End-Functionalized Polymers/Oligomers by Olefin Metathesis Reactions
Conjugated
AUTHOR : Abdellatif, Mohamed Mehawed
EXAMINED BY
Examiner in chief
Examiner
Examiner
Examiner
QUALIFIED BY THE GRADUATE SCHOOL
OF SCIENCE AND ENGINEERING
TOKYO METROPOLITAN UNIVERSITY
Dean
Date
cd,„
1G~rLi Z~, Zv l 3
Chapter 1
General Introduction
Contents
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
n-Conjugated Polymers
Conventional Synthetic Routes for n-Conjugated Polymers Olefin Metathesis
Controlled Radical Polymerization Click Chemistry
Contribution of Our Research Group Purpose on This Work
References
1-1. a-Conjugated Polymers
Since the initial discovery in 19701, that polyacetylene (CH)X, now commonly known as the prototype conducting polymer, could be p- or n-doped either chemically or electrochemically to the metallic state, the development of the field of conducting polymers has continued to accelerate at an unexpectedly rapid rate and a variety of other conducting polymers and their derivatives have been discovered.2
7E-Conjugated polymer can be defined as an organic polymer that possesses the electrical, electronic, magnetic, and optical properties of a metal while retaining the mechanical properties, processibility, etc. commonly associated with a conventional polymer, is termed an "intrinsically conducting polymer" (ICP) more commonly known as a "synthetic metal", and their conductivity can be changed by several orders of magnitude from a semiconducting state to a metallic state by doping. Usually p-doping is achieved by partial oxidation of the polymer by a chemical oxidant or an electrochemical method, and causes depopulation of the bonding it orbital (HOMO) with the formation of "holes".2
\R R
N ~ S
/nnn
polyacetylene polyaniline polyphenylenepolythiophene
n
poly(p-phenylene vinylene)
\ / \ /\ • /
R nR R
polycarbazolepolyfluorene
Figure 1-1. n-Conjugated Polymers.
n
Conducting polymers (and most of their derivatives) (Figure 1-1), undergo either p- and/or n-redox doping by chemical and/or electrochemical processes during which the number of
electrons associated with the polymer backbone changes.3'4
n-Conjugated polymer combining mechanical flexibility, and ease in band-gap /color-tuning
via structural control, along with the potential for low-cost scalability and processing, are attractive in the fast-growing area of plastic electronics. For instance, in considering the most established applications, polymeric semiconductors are expected to find widespread application in thin film transistors (OTFTs),5'6 photovoltaic cells,7'8 radiofrequency identification (RFID) tags, sensors,9 memories,10'11 and light emitting diodes (OLEDs).12
1-2. Conventional Synthetic Routes for it-Conjugated Polymers 1-2-1. Condensation under high temperature in vacuo
In this process, treatment of the bis(sulfonium halide) salt of p-xylene with 1 equiv of base induces a 1,6-elimination to generate the quinodimethane-like intermediate, which subsequently undergoes 1,6-polymerization to produce the sulfonium precursor polymer. This water-soluble precursor can then be spin-cast into a thin film and converted in situ into conjugated poly(p-phenylenevinylene) after thermal treatment in a vacuum to trigger a 1,2-elimination (Scheme 1-1).13
The drawback of this methodology is by-production of halogen (inorganic halide, sulfur) which should damage the property and additional cost needed for purification. Moreover, carbonyl moieties are present as defects, formed during the thermal conversion of a precursor to poly(p-phenylenevinylene) (PPV), a polymer used in light emitting diodes. The increase in carbonyl groups can be correlated with a dramatic reduction of PPV photoluminescence.14
CHCICI-~ 2~S SC + Base---- CIS S+1„,
CIH2CMcOHCr
CI $+
~®heat 250°C vacuum ®+ \2HCI
n
Scheme 1-1. Synthesis of PPV via quinodimethane intermediate
1-2-2. Gilch polymerization
Condensation in the presence of inorganic salt, which started similarly to various precursor
3
methods (cf. sulfonium precursor: Wessling route; chloro precursor route; xanthogenate precursor route; sulfinyl precursor route), is the base-induced elimination of HC1 which leads to the "real monomer", a quinodimethane derivative (Scheme 1-2). For the second steps the polymerization itself, there are two reasonable possibilities: (i) a free radical mechanism, which is claimed for precursor polymerization (ii) an anionic induced polymerization. It is even possible that these two pathways compete with each other, depending on the exact reaction conditions. The third (and last) step is the polymer analogous elimination of HC1 by a second equivalent of base.15
R RR R
CI
40. R= alkyl etc.n KOtBu 01.+CI2KC1
Scheme 1-2. Synthesis of PFV by Gilch Polymerization
However, Gilch route suffers from the generation of polymers having saturated defects along the chain backbone (predominantly tolane-bisbenzyl-type moieties analogous to those commonly found in samples of PPV).16
1-2-3. Horner-Emmons (HWE) polymerization
Poly(9,9-dialkylfluorenyl-2,7-vinylenes) (PFVs) having high molecular weights and no detectable saturated defects along the conjugated backbone has been prepared by coupling
suitably designed comonomers to form the targeted conjugated polymers (Scheme 1-3). However, the drawback of this methodology is too many steps (7 steps) needed to synthesize the corresponding biphosphate monomer. 17
O
OHC4%0CHO + (EtO)92P4t4P(OEt)2
R R R= alkyl etc . R R
KOtBu
\ •
R R
Scheme 1-3. Synthesis of PFV by Horner- Emmons Polymerization
4
1-2-4. Transition-metal-catalyzed • cross-coupling reactions
The reaction, in general, involves a transition-metal-catalyzed oxidative addition reaction across the C-X bond of an electrophile and then transmetalation with a main group organometallic nucleophile, followed by a reductive elimination step leading to the carbon-carbon bond formation. Concomitantly the active catalyst is regenerated. The most commonly employed transition-metal catalysts are nickel- or palladium based complexes, although other metals have also been used. The organometallic nucleophiles can be Grignard reagents (Kumada-Corriu),18 stannyl (Stille),19 boron reagents (Suzuki-Miyaura),20 or copper (Sonogashira).21
Suzuki couplingKumada coupling
2eq K2CO3 aqNi(dppb)Cl2 or
3 mol- %Pd(PPh3)4/R'X + RMgX> R'-R
B(OH)2 + Br_®Pd(PPH3)4
iRbenzene, heat— R
R = Aryl, Vinyl, Alkyl
R' = Aryl, Vinyl X = CI > Br >
Sonogashira coupling
(Ph3P)2PdCl2
Scheme 1-4. Transition-metal-catalyzed cross-coupling reactions 1-3. Olefin Metathesis
Olefin metathesis is a powerful synthetic technique in forming carbon—carbon bo Using this mild reaction in forming carbon—carbon double bonds, complex reaction (Figure 1-2) have developed beyond the simplest olefin metathesis mode involving ti metathesis (CM).23 CM involves two different olefins and heterointerchange, self-metathesis (SM) involves two of the same olefins and homointerchange as a
Furthermore, rings may be opened using ROM25 with a selected opening species or Olefin metathesis is a powerful synthetic technique in forming carbon—carbon nds.22'23 Using using this mild reaction in forming carbon—carbon double bonds, complex reaction modes (Figure 1-2) have developed beyond the simplest olefin metathesis mode involving the cross metathesis (CM).23 CM involves two different olefins and heterointerchange, whereas self-metathesis (SM) involves two of the same olefins and homointerchange as a result.24 Furthermore, rings may be opened using ROM25 with a selected opening species or dos(
ringclosing metathesis (RCM)26'27 releasing ethylene. A combination of metathesis modes coupled
with understanding reactivity has even allowed for metathesis polymerization modes involving
either chain polymerization (ROMP)25'28 or step polymerization acyclic diene metathesis
(ADMET).29
X( n )
RCMADMET
Ring-ClosingAcyclic Diene
Metathesis
,G~° C:Dy Metathesis
nX ROMP(X
--- Ring-Opening (\)
Metathesis\n
Polymerization
2- CM R1+R R1 R2
Figure 1-2. Modes of metathesis
1-3-1. Metathesis catalyst and initiator evolution
It was the developments of the 1970s by Richard Schrock, then a researcher at DuPont, toward homogenous metathesis catalysts that eventually led to the isolation of a high oxidation state nucleophilic carbenes, termed "Schrock carbenes," including tantalum carbene complexes.3°
Schrock carbene ligands are viewed as X2 ligands with +2 charge in contrast to the neutral Fischer carbene L ligands. With tantalum catalyst activity too low, further catalyst developments using tungsten31'32 and molybdenum33'34 followed (Chart 1-1).
The multitude of highly active early-transition metal catalysts allowed for diverse applications that were limited by the electrophilic and oxophilic metal centers sensitive to air, moisture, and functional groups. Late-transition metals remained desirable as they are less electrophilic and moving toward that direction. Schrock and Toreki eventually also created olefin metathesis active rhenium complexes.35 From Schrock's work, numerous achievements in small molecules and polymers were possible.
Simultaneous discoveries by Grubbs and coworkers throughout the 1980s led to
spectroscopically characterizable titanacyclobutanes from titanium/Lewis acid mixtures,36-38
which brought the understanding of olefin metathesis to a more complete level. Incidentally, the
Wagener research group was just being established at this time in 1984. Grubbs and coworkers
also found success on moving from the titanium/ Lewis acid mixtures to well-defined
homogenous catalysts with rhenium and the more successful ruthenium centers such with Grubbs'
first-generation catalyst (Rul) and second-generation catalyst (Ru2) (Chart 1-1).40-41
Although ruthenium has proven to be the most successful late-transition metal in reducing air, moisture, and coordinating functionality sensitivity, the first-generation catalysts were less active than early-transition metal catalysts. In particular, structures such as those shown in (Chart 1-1) were interesting in that they exhibited features of Fischer and Schrock carbenes, but not exclusively classified as either. This allowed for a continuum between early- and late-transition metals, dependent on metal and ligand selection in altering electronics and/or sterics for particular metathesis modes.
Certainly, all of these developments have established a huge library of catalysts from which a number of synthetic challenges in pharmaceuticals, petrochemicals, and polymers have been met.
These catalyst developments have also allowed for a multitude of developments in the more specialized ADMET polymerization reaction since the early 1990s.
i'1'
„..-.r.,„,- ,r-„
N N
iiu ROIW—ROIMo
ROIR^ -)(
H3C CH3H3C (
•
W1Mol
R = CCH3(CF3)2
Ar—NYN-Ar CI,,,PCy3 CI,,,I • R
u—CHPhRu CHPh
II
PCy31PCy31 RulRu2
Ar = 2,4,6-Me3C6H2, Cy = cyclohexyl
Chart 1-1. Catalysts for ADMET
1-3-2. Acyclic Diene Metathesis (ADMET) polymerization
As ADMET is classified as a condensation polymerization, and as with most condensation
polymerizations, this equilibrium process requires desired functional groups reacting in a
step-wise fashion allowing for molecular weight increase proceeding stepwise from monomer,
dimer, trimer, and so on to high polymer.42 This stepwise event occurs as the linearly coupled
molecules are released during the metathesis catalytic cycle. To drive the equilibrium, ethylene is
removed from the reaction. At high conversion, each coupling between polymer chains reduces
the number of polymer molecules in the sample significantly. This stark contrast to the situation at
low conversions, which results in only a small reduction of polymer molecules, results in a low
polymerization rate until high conversions are realized. Implications on catalysis include
necessitating a highly active catalyst that remains active throughout the polymerization cycle.
High conversions are achieved by removing the condensate, driving the equilibrium. Using the least sterically demanding monomers, a,co-dienes, the ethylene condensate is removed, thus preventing the reverse reaction regenerating monomer. Despite the high conversion required for high molecular weight as with typical condensation polymers, several commercially important condensation polymers are used successfully including polyesters and polyamides.
Molecular weight distributions in ADMET polymers are also typical of other condensation polymers. The polydispersityindex (PDI) is defined as the weight-average molecular weight, X„,,
divided by the number-average molecular weight, Xn, and describes the molecular weight distribution. Statistically, condensation polymers typically have PDIs approaching 2.0, the most probable distribution, as conversion reaches 1 or 100%.29
uicuiyiiuciic carbine (he atrue catalyst in this step polymerization). The open coordination site allows for olefin coordination, and 2 + 2 cycloaddition forming a metallocyclobutane,which decomposes by a 2 + 2 cycloreversion, releasing ethylene condensate and resulting in an a-substituted metal alkylidene species may be monomer polymer molecule productively 1-3-2-1. ADMET mechanism
The polymerization cycle proceeds through the same basic metal—carbene mechanism as in CM with features optimized to enhance polymerization through productive metathesis (Scheme
44-461
-5) .43, 446Precatalyst
The precatalyst involves a metal _1~
species with associated ligands [M],R~[M]=CH2
polymer \~
which reacts reversibly to form a metalR-/\EM]
J
release of ethylene
= diene (a monomer or polymer of any size [M] = metal with associated ligands
Scheme 1-5. Productive ADMET polymerization cycle alkylidene with an open coordination site. The metal
orpolymer, and coordination of another monomer or suli
twolinearlycoupledmoleculesandregeneratesthemetal
I-~K
R~
methylidene carbene.
The cis:trans stereochemical outcome of productive ADMET with Grubbs catalysts has implications on the overall polymer properties yet is rooted in the mechanism. With cis:trans ratios of 3:7 and 1:4 with more time, the preference of trans can be rationalized with the lesser sterically favored approach and resulting a,fl-metallocyclobutane conformation (Scheme 1-6).47
,CI Ru=HL
CrR [Ru]
RRu=CH2 +
RR CI
Scheme 1-6. Stereochemical trans outcome rationale 1-4. Controlled Radical Polymerization
Worldwide production of synthetic polymers is in the range of 200,000,000 tonnes per year, or approximately 40 kg per person, half of this is based on copolymers prepared by radical polymerization (RP) — a chain polymerization in which the reactive centres are radicals. Until recently, polymers prepared by RP were mainly used as commodity plastics, rubbers and fibres because of an inadequate level of control over molecular structure. Many high-value applications require well-defined polymers, in terms of composition and molecular architecture, which, for a
long time, could only be prepared via living (ionic) polymerization.48 Living polymerization — where chain-breaking reactions such as termination and transfer are absent and all chains are instantaneously initiated and grow simultaneously — enables chemists to create polymers with precisely controllable molecular weights and narrow molecular weight distributions, and also determines every aspect of macromolecular architecture (composition, topology and functionality).49
1-4-1. Atom Transfer Radical Polymerization (ATRP)
Atom transfer radical polymerization (ATRP) is the most extensively studied
controlled/living radical polymerization (CRP) method, with the interest originating primarily in
its simplicity and broad applicability, and in the ability to prepare previously inaccessible
well-defined nanostructured polymeric materials.49
ATRP is a catalytic process and can be mediated by numerous redox-active transition metal complexes (Cu has been the most often used transition metal but other studied metals include Ru, Fe, Mo, Os).49
kactR-XX X XX X X XX
R-X + LCu'Y --R • + LCu°(X)Y X XX
X ""!... X
kdeact
RX X ^~~/X""""""X
(c)i.XK X XX Grafting fromX XX"
monomer t
erminationXxaft
Scheme 1-7. Atom transfer radical polymerization
In ATRP,alkyl halide initiators or dormant species (RX) react with activators — low-oxidation-state metal complexes LCuIY, to reversibly form both propagating radicals (R), and deactivators — higher-oxidation-state metal complexes with coordinated halide ligands. The dormant species in this ATRP equilibrium can be polymer chains able to grow in one or many directions, or polymers attached to functional colloidal particles, surfaces, biomolecules and so on (Scheme 1-7).49
functional polymer
Grafting to {J4litrtilajW0.•
telechelic polymer monomar R'
Grafting frommacroinitiator
srr star shape
Ph3P -NHITMS ~zO
-NHZ
+DBU
OHTiCl4CNH
OH0412:PRO
Figure 1-3. Examples of controlled macromolecular architecture ATRP and chemical modofications at the chain end.
CuBr / L
.,uBr / L
•.
X
0
X OH
in polymers prepared by
10
ATRP permits the synthesis of (co)polymers with targeted composition, controlled molecular architecture51, predetermined MW and narrow MWD, which makes this method very attractive for industry52. The chain topologies include statistical, gradient and segmented (block or graft) copolymers and for each of these cases, the chain architecture can be varied in a controlled manner, to include combs, brushes, multiarm stars and dendritic macromolecules with controlled degrees of branching. In contrast to conventional RP - where the polymer chains grow and terminate within a very short time period (<1 s), in CRP, though a small fraction do still terminate, most chains grow throughout the duration of the polymerization (>1 h). As a result, in copolymerization, any changes in the instantaneous copolymer composition as a function of monomer conversion in CRP are observed along the backbone of each individual chain. Therefore, the copolymer chains not only have nearly identical MW but also the same compositional gradient along the backbone. Such gradient or tapered copolymers are not readily accessible by other polymerization techniques53. The polymers prepared by ATRP contain a terminal radically transferable atom — usually a halogen — and can participate in various post-polymerization reactions54, or serve as macroinitiators in the synthesis of block copolymers51. The use of functional ATRP initiators and the ease with which alkyl halides can be attached to complex structures such as natural products, organic or inorganic particles and surfaces further expands the number of accessible materials to various nanocomposites55 and (bio)degradable functional macromolecules (Figure 1-3).50
1-5. Click Chemistry
Click chemistry encompasses a wide range of reactions that have proven to be powerful and versatile additions to the chemist's toolbox. The broad utility of this class of reactions stems from its inherent simplicity and efficiency. Click reactions are characterized by selectivity, facile experimental set-up, applicability in aqueous and aerobic systems, tolerance to a variety of functional groups, quantitative yields, and minimal synthetic work-up.56 Several reactions exhibit these features, including Cul-catalyzed azide–alkyne cycloaddition,57 Diels–Alder
cycloaddition,58'59 the thiol-ene reaction,60 and azide–nitrile cycloaddition.61 Of these reactions,
the CuI-catalyzed azide–alkyne cycloaddition dominates the recent body of literature on click
chemistry.
These simple and versatile reactions can greatly facilitate the synthesis and modification of polymeric materials.62 There is a wide range of diverse materials that has been prepared in conjunction with various click reactions, including: terminal- and pendant-functional polymers;
(multi)block copolymers and micelles; complex architectures such as star, brush, graft, hyperbranched, and dendritic polymers; gels and networks; and polymers conjugated to nanomaterials. Many of the polymers that are used for furthermodifications were originally prepared by controlled polymerization techniques. Controlled radical polymerization (CRP) allows for the preparation of polymers with a pre-determined molecular weight, narrow molecular weight distribution, chain end functionality, and tightly controlled architecture and composition.63 The most commonly employed CRP techniques include atom transfer radical polymerization (ATRP),64 reversible addition-fragmentation transfer (RAFT) polymerization,65 and stable free radical polymerization (such as nitroxide-mediated polymerization, NMP).66,67
1-5-1. CuI-catalyzed huisgen 1,3-dipolarcycloaddition of azides and terminal alkynes
The CuI-catalyzed Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes to form 1,2,3-triazoles is the model example of a click reaction. It fulfills all of the criteria of click chemistry perfectly, no matter how subjective they may be, and is therefore extremely reliable and easy to use. This reaction exclusively forms 1,4-substituted products, making it regiospecific.
It typically does not require temperature elevation but can be performed over a wide range of temperatures (0-160°C), in a variety of solvents (including water), and over a wide range of pH values (5 through 12). It proceeds as much as 107 times faster than the uncatalyzed version, and purification essentially consists of productfiltration69°7°-72. Furthermore, it is unaffected by steric factors. "Variously substituted primary, secondary, tertiary, and aromatic azides readily participate in this transformation. Tolerance for variations in the acetylene component is also excellent"71. All of these characteristics make this cycloaddition particularly popular among the other click reactions mentioned previously.
Two additional reasons for the popularity of this cycloaddition are azides and terminal
alkynes are fairly easy to install and they are extremely stable at standard conditions68'73. They
both can tolerate oxygen, water, common organic synthesis conditions, biological molecules, a large range of solvents and pH's, and the reaction conditions of living systems (reducing environment, hydrolysis, etc.)68°69,74Even though the decomposition of aliphatic azides is thermodynamically favored, a kinetic barrier exists that allows them to be stable in the aforementioned conditions69. They will essentially remain "invisible" in solution until a dipolarophile, such as an alkyne, comes into contact69.
1-5-1-1. Mechanism of CuI-catalyzed Huisgen 1,3-dipolar cycloaddition (HDC)
In general, cycloadditions proceed through a concerted mechanism. However, experimental kinetic data75 and molecular modeling72 performed on the HDC reaction seem to favor a stepwise reaction pathway69'70
Based on experimental evidence70',71 and the fact that Cu' can readily insert itself into
terminal alkynes [Sonogashira coupling76], it is envisioned that the first step of the reaction
involves 7C complexation of a Cu' dimer to the alkyne (1 in Scheme 1-8). Thereafter, deprotonation
of the terminal hydrogen occurs to form a Cu-acetylide70. There are actually several different
kinds of Cu-acetylide complexes that can form, depending on the reaction conditions utilized; 2
represents just one possibility72. The 7C complexation of Cu' lowers the pKa of the terminal alkyne
by as much as 9.8 pH units, allowing deprotonation to occur in an aqueous solvent without the
addition of a base69. If a non-basic solvent such as acetonitrile was to be used, then a base, such as
2,6-lutidine or N,N'-diisopropylethylamine (DIPEA), would have to be added77.
,L\ R1--- H 2CuL
~-Cu/Cu --- L
Cu Catalyst
R2~N,N'~N
H Ri
B'5
R1 R1
L
Cu Cu
1 g- B-H
/L• ---C u'
L 2
R2-N3 B-H
RZ-_N~N`~N
~C u)-(I — Metallocycle,•—
3 „N I ,N~ R 2
R1
`_ 4
Cu Cu L
L 3 Cu
Scheme 1-8. Proposed mechanism for the HDC reaction.
In the following step, N(1) displaces one of the ligands from the second Cu in the Cu-acetylide complex to form 3. In turn, this "activates" the azide for nucleophilic attack C(5).
Due to proximity and electronic factors, N(3) can now easily attack C(4) of the alkyne, leading to a metallocycle (not shown for simplicity). The metallocycle then contracts when the lone pair of electrons of N(1) attacks C(5) to form the respective triazole 4. Once 4 forms, the attached Cu dimer immediately complexes to a second terminal alkyne. However, this second alkyne cannot undergo a cycloaddition due to the unfavorable structure of the complex, and it dissociates upon protonation to reform 4. One final protonation releases the CuI catalyst from the 1,2,3 -triazole product 5, to undergo a second catalytic cycle with different substrates69. Both of these protonations are most likely the result of interactions with protonated external base and/or solvent, but further studies are needed to conclusively confirm.69'78
14
1-6. Contribution of Our Research Group
Nomura et al. recently demonstrated syntheses of defect-free, stereo-regular (all-trans), high molecular weight poly(9,9-dialkylfluorene-2,7-vinylene)s (PFVs),79 poly(2,5- dialkylphenylene-1,4-vinylene)s (PPVs),80 poly(N-alkylcarbazole-2,7-vinylene)s79 by acyclic diene metathesis (ADMET) polymerization. Since the resultant polymers prepared by the Ru-carbene catalyst possess well-defined polymer chain ends (as vinyl group),79,8° a facile, exclusive end-functionalization can be achieved by treating the vinyl groups with the Mo-alkylidene(Mocat.)followedbyWittig-typecleavaewith Y()YgYY aldehde.79,8'Precise syntheses of ABA type amphiphilic triblock copolymers (Scheme 1-9),79 and of PFV's containing oligo- (thiophene)s in the both chain ends which exhibit unique emission properties by an energy transfer (Scheme 1-10) have been demnostrated.79
R R
\ \Ru cat.°•,~.\ R R
n 10. ---
°.R Rn-1 ip
R = n-octylPFV
+ (n-1) H2C=CH2
//-\® 1 i) Mo cat . (5 equiv.)
Ar—NYN-Ar
CI,,,
`NoMo°°
Pcy31(F3C)2MeCO`j°'CHCMe2Ph\11*iik(F ICI46,--®R 3C)2MeCO•°°° R
Ru cat.Mo cat.R Rn-i;Mo';
Ar = 2,4,6-Me3C6H2,
Cy = cyclohexyl1 ii) 4-Me3SiO-C6H4CHO (excess)
/\ 7\ p p E~nFunctionalization
•
Me3SiO\ \
i) 0.5 N HCI aq.RR ii) KH in THE
Ms = McSO2 iii) Ms0"("\--- ");ms PEGMs2
Mst0\ ,
O 4e.
PEG-b/-PFV-b/-PEGR R
Scheme 1-9. Synthesis of ami
n-1
R R ="
PFV-OTMS
R R
•e e
n-1
f amphiphilic triblock copolymers
O OSiMe3
I-Ms
15
R R
\ 40.Ru cat. }4
R = n-octyl Ar—N Y N-Ar
CI,, I Ru CHPh
PCy3 I (F3C)2MeCO`I o'CHCMe2Ph
(F3C)2MeCO
R u cat.Mo cat.
Ar = 2,4,6-Me3C6H2, Cy = cyclohexyl
MesSiO, ®~.
i) 0.5 N HCI aq.
ii) KH in THE
Ms = McS02)MsO"N'0)R,N Ms(—1
40
PEG-b/-PFV-b/-PEG
, .
PFV
+ (n-1) H2C=CH2
i) Mo cat. (5 equiv.)
Mo
R R
iii) MsO"~(3)r„Ms PEGMs2
\ \
MeO OMe
5 \
OMe
ii)`7,>CHO (excess)
—CHO MP3T-CHO
- [Mo=O]
R Wittig-type cleavage Mo /R Mo0=\—.-O CHR'
R'
PEG-b/-PFV-b/-PEG \ R' R \ , / \ / \ ••:s• \ / O1 0.4_Ms
Scheme 1-10. Synthesis of poly(fluorene-2,7-vinylene)s containing oligo- (thiophene)s at the chain ends
Optically active aggregates in the chiral tersolvents µm-sized polymer particles from achiral all-trans-poly(9,9-di-n-octylfluorene-2,7-vinylene) (PFV) have been succfully attained. This work demonstrated through collabrotive research with Fujiki et al., where emergence and enhancement of chiroptical signals of µm-sized polymer particles from achiral PFV during mirror-symmetry-breaking aggregation due to the optically active fluidic media consisting of (R)-limonene and (S)-limonene, chloroform and methanol. These results were proven by refractive index and specific rotation of the media as well as circular dichroism (CD), optical rotatory dispersion, circular polarised luminescence (CPL), UV-vis and photoluminescence spectral characteristics of PFV. For comparison, a triple bond linker poly[(9,9-di-n-octylfluoren-2,7-diyl)- alt-yleneethynylene] (PFE) aggregate (Chart 1-2), which is an analogue of PFV, did not show any CD-signals in the 71-n* transition. Gaussian 03 (TD-DFT, B3LYP, 3-21G basis set) calculations of PFV and PFE trimer models suggested that PFV is CD-/CPL-silent helix due to an equal proportion of P- and M-helices in a double-well with a small barrier height, conversely, that PFE is inherently optically inactive due to non-helix conformation in a single-well. 82
16
17C8H17 PFV
CH3
CH3 (R)-(+)-Iimonene
CH3
%OP
CHCHH
817817 ,CH3
PFE (S)-(+)-Iimonene
Chart 1-2. Chemical structures of PFV, PFE and limonenes used
The previous demonstrations show that the defect-free, stereo-regular (all-trans), high
molecular weight poly(9,9-di-n-octylfluorene-2,7-vinylene) synthesized by ADMET
polymerization technique is a promising candidate for optical devices because it emits blue light
at a nearly 100% On in solution at room temperature;79 in addition to, the emergence and
enhancement of chiroptical signals in the optically active fluidic media.
1-7. Purpose on This Work
This thesis consists of two chapters with the theme of olefin metathesis reactions using transition metal catalysts to synthesize end-functionalized conjugated polymers/oligomers in precise manner.
One is preparation of various block (graft) copolymers by combination of ADMET technique with Cu catalyzed atom transfer radical polymerization (ATRP) technique by using macroinitiators prepared by introductions of the initiating functionalities into PFV chain ends (grafting from approach). Moreover, amphiphilic ABCBA type block copolymers containing PEG fragment has been attained by additional combination with click chemistry (grafting to approach).
The precise control of the amphiphilic nature as well as of the block lengths via synthesis shall open the way to fine-tuning the lateral dimensions of these nanostructures. The facts should be thus highly promising for designing materials for the desired purposes.
In chapter 3, a possibility to establish a synthetic methodology for preparation of new conjugated materials with unique optical properties has been explored. Through this study, the methodology for synthesis of end-functionalized oligo(2,5-dialkoxy-phenylene-1,4-vinylene)s
(OPVs) with exact repeating units has been extended by adopting a coupled olefin metathesis
with Wittig-type coupling. A wide applicability of this methodology enables us to fine tuning of
various conjugated materials (oligomer, polymers) with unique properties.
1-8. References
(1) Ito, T.; Shirakawa, H.; Ikeda, S. .1 Polym. Sci. Polym. Chem. Ed. 1974, 12, 11.
(2) MacDiarmid, A.G. Angew. Chem. Mt. Ed., 2001, 40, 2581.
(3) Handbook of Conducting Polymers, Vol 1&2 (Ed.: T. A. Skotheim), Marcel Dekker, New York, 1986.
(4) M. G. Kanatzidis, Chem. Eng. News 1990, 68(49), 36.
(5) Zaumseil, J.; Sirringhaus, H. Chem. Rev. 2007, 107, 1296.
(6) Allard, S.; Forster, M.; Souharce, B.; Thiem, H.; Scherf, U. Angew. Chem., Mt. Ed. 2008, 47, 4070.
(7) Thompson, B. C.; Frechet, J. M. J. Angew. Chem., Mt. Ed. 2008, 47, 58.
(8) Gunes, S.; Neugebauer, H.; Sariciftci, N. S. Chem. Rev. 2007, 107, 1324.
(9) Thomas, S. W.; Joly, G. D.; Swager, T. M. Chem. Rev. 2007, 107, 1339.
(10) Moller, S.; Perlov, C.; Jackson, W.; Taussig, C.; Forrest, S. R. Nature 2003, 426, 166.
(11) Ling, Q.-D.; Song, Y.; Lim, S.-L.; Teo, E. Y.-H.; Tan, Y.-P.; Zhu, C.; Chan, D. S. H.;
Kwong, D.-L.; Kang, E.-T.; Neoh, K.-G. Angew. Chem., Mt. Ed. 2006, 45, 2947.
(12) Kulkarni, A. P.; Tonzola, C. J.; Babel, A.; Jenekhe, S. A. Chem. Mater. 2004, 16, 4556.
(13) van Breemen, A. J. J. M.; Issaris, A. C. J.; de Kok, M. M.; Van Der Borght, M. J. A.;
Adriaensens, N. P. J.; Gelan, J. M. J. V.; Vanderzande, D. J. M. Macromolecules 1999, 32, 5728.
(14) Papadimitrakopoulos, F.; Konstadinidis, K.; Miller, T. M.; Opila, R.; Chandross, E. A.;
Galvin, M. E. Chem. Mater. 1994, 6, 1563.
(15) Becker, H.; Spreitzer, H.; Ibrom, K.; Kreuder, W. Macromolecules 1999, 32, 4925.
(16) Becker, H.; Spreitzer, H.; Kreuder, W.; Kluge, E.; Schenk, H.; Parker, I.; Cao, Y. Adv. Mater.
2000, 12, 42.
(17) Anuragudom, P.; Newaz, S. S.; Phanichphant, S.; Lee, T. R. Macromolecules 2006, 39, 3494.
(18) Tamao, K.; Sumitani, K.; Kumda, M. J Am. Chem. Soc. 1972, 94, 4374.
(19) Stille, J. K. Angew. Chem., Int. Ed. 1986, 25, 508.
(20) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(21) Sonogashira, K. J. Organomet. Chem. 2002, 653, 46.
(22) Ivin, K. J.; Mol, J. C. Olefin Metathesis and Metathesis Polymerization, 2nd ed.; Academic Press: San Diego, CA, 1997; pp 1-472.
(23) Hoveyeda, A. H.; Zhugralin, A. R. Nature 2007, 450, 243.
(24) Basset, J.-M.; Coperet, C.; Soulivong, D.; Taoufik, M.; Cazet, J. T. Acc. Chem. Res. 2009, 43, 323.
(25) Bielawski, C. W.; Grubbs, R. H. Prog. Polym. Sci. 2007, 32, 1.
(26) Dieters, A.; Martin, S. F. Chem. Rev. 2004, 104, 2199.
(27) McReynolds, M. D.; Dougherty, J. M.; Hanson, P. R. Chem. Rev. 2004, 104, 2239.
(28) Buchmeiser, M. R. Chem. Rev. 2000, 100, 1565.
(29) Opper, K. L.; Wagener, K. B. J Polym. Sci. Part A: Polym. Chem. 2011, 49, 821.
(30) Schrock, R. R..1 Am. Chem. Soc. 1974, 96, 6796.
(31) Schaverien, C. J.; Dewan, J. C.; Schrock, R. R. J. Am. Chem. Soc. 1986, 108, 2771.
(32) Schrock, R. R.; DePue, R. T.; Feldman, J.; Schaverien, C. J.; Dewan, J. C.; Liu, A. H. I Am.
Chem. Soc. 1988, 110, 1423.
(33) Schrock, R. R.; Murdzek, J. S.; Bazan, G. C.; Robbins, J.; DiMarie, M.; O'Regan, M. J. Am.
Chem. Soc. 1990, 112, 3 875 .
(34) Oskam, J. H.; Fox, H. H.; Yap, K. B.; McConville, D. H.; O'Dell, R.; Lichtenstein, B. J.;
Schrock, R. R. .1 Organomet. Chem. 1993, 459, 185.
(35) Schrock, R. R.; Toreki, R..1 Am. Chem. Soc. 1990, 112, 2448.
(36) Howard, T. R.; Lee, J. B.; Grubbs, R. H. J. Am. Chem. Soc. 1980, 102, 6876.
(37) Lee, J. B.; Ott, K. C.; Grubbs, R. H. J. Am. Chem. Soc. 1982, 104, 7491.
(38) Gilliom, L. R.; Grubbs, R. H. J. Am. Chem. Soc. 1986, 108, 733.
(39) Nguyen, S. T.; Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1992, 114, 3974.
(40) Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc. 1993, 115, 9858.
(41) Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1995, 117, 5503.
(42) Odian, G. Principles of Polymerization, 4th ed.; Wiley Interscience: Hoboken, NJ, 2004, pp 39-350.
(43)Wagener, K. B.; Boncella, J. M.; Nel, J. G. Macromolecules 1991, 24, 2649.
(44) Wagener, K. B.; Nel, J. G.; Konzelman, J.; Boncella, J. M. Macromolecules 1990, 23, 5155.
(45) Wagener, K. B.; Boncella, J. M.; Nel, J. G.; Duttweiler, R. P.; Hillmyer, M. A. Makromol.
Chem. 1990, 191, 365.
(46) Wagener, K. B.; Nel, J. G.; Duttweiler, R. P.; Hillmyer, M. A.; Boncella, J. M.; Konzelman, J.; Smith, D. W., Jr.; Puts, R.; Willoughby, L. Rubber Chem. Technol. 1991, 64, 83.
(47) Konzelman, J. Ph.D. dissertation, University of Florida, 1993.
(48) Szwarc, M. Nature 1956, 176, 1168.
(49) Matyjaszewski, K.; Tsarevsky, N. V. Nature Chemistry 2009, 1, 276.
(50) Tsarevsky, N. V.; Matyjaszewski, K. Chem. Rev. 2007, 107, 2270.
(51) Davis, K. A.; Matyjaszewski, K. Adv. Polym. Sci. 2002, 159, 1.
(52) Matyjaszewski, K.; Spanswick, J. Mater. Today 2005, 8, 26.
(53) Matyjaszewski, K.; Ziegler, M. J.; Arehart, S. V.; Greszta, D.; Pakula, T. J. Phys. Org.
Chem. 2000, 13, 775.
(54) Coessens, V.; Pintauer, T.; Matyjaszewski, K. Prog. Polym. Sci. 2001, 26, 337.
(55) Pyun, J. & Matyjaszewski, K. Chem. Mater. 2001, 13, 3436.
(56) Kolb, H. C.; Finn M. G.; Sharpless, K. B. Angew. Chem., Mt. Ed. 2001, 40, 2004.
(57) Huisgen, R. 1,3-Dipolar Cycloaddition Chemistry, Wiley, 1984; Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Mt. Ed. 2002, 41, 2596; Tornoe, C. W.;
Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057.
(58) Durmaz, H.; Dag, A.; Altintas, 0.; Erdogan, T.; Hizal, G.; Tunca, U. Macromolecules 2007, 40, 191.
(59) Kim, T.-D.; Luo, J.; Tian, Y.; Ka, J.-W.; Tucker, N. M.; Haller, M.; Kang, J.-W.; Jen, A. K.
Y. Macromolecules 2006, 39, 1676.
(60) Becer, C. R.; Hoogenboom, R.; Schubert, U. S. Angew. Chem., Mt. Ed. 2009, 48, 4900;
Dondoni, A. Angew. Chem., Int. Ed. 2008, 47, 8995; Killops, K. L.; Campos, L. M.; Hawker, C. J.
J Am. Chem. Soc. 2008, 130, 5062.
(61) Demko, Z. P.; Sharpless, K. B. J Org. Chem. 2001, 66, 7945; Tsarevsky, N. V., Bernaerts, K. V. ; Dufour, B.; Du Prez, F. E. Matyjaszewski, K. Macromolecules, 2004, 37, 9308.
(62) Golas, P. L.; Matyjaszewski, K. QSAR Comb. Sci. 2007, 26, 1116; Binder, W. H.;
Sachsenhofer, R. Macromol. Rapid Commun. 2007, 28, 15; Binder, W. H.; Sachsenhofer, R.
Macromol. Rapid Commun. 2008, 29, 952; Lundberg, P.; Hawker, C. J.; Hult, A.; Malkoch, M.
Macromol. Rapid Commun. 2008, 29, 998.
(63) Braunecker, W. A.; Matyjaszewski, K. Prog. Polym. Sci. 2007, 32, 93; Matyjaszewski, K.;
Braunecker, W. A. In Radical Polymerization, ed. Matyjaszewski, K.; Gnanou, Y.; Leibler, L.
Wiley-VCH, Weinheim, Germany, 2007; Matyjaszewski, K.; Davis, T. P. In Handbook of Radical Polymerization, Wiley, Hoboken, 2002.
(64) Kamigaito, M.; Ando, T.; Sawamoto, M. Chem. Rev. 2001, 101, 3689; Matyjaszewski, K.;
Xia, J. Chem. Rev. 2001, 101, 2921; Wang, J.-S.; Matyjaszewski, K. J. Am. Chem. Soc. 1995, 117, 5614; Matyjaszewski, K.; Tsarevsky, N. V. Nat. Chem. 2009, 1, 276; Tsarevsky, N. V.;
Matyjaszewski, K. Chem. Rev. 2007, 107, 2270.
(65) Moad, G.; Chiefari, J.; Chong, Y. K.; Krstina, J.; Mayadunne, R. T. A.; Postma, A.;
Rizzardo, E.; Thang, S. H. Polym. Mt. 2000, 49, 993; Rizzardo, E.; Chiefari, J.; Mayadunne, R.;
Moad, G.; Thang, S. Macromol. Symp. 2001, 174, 209; Cunningham, M. F. Prog. Polym. Sci.
2008, 33, 365; Lowe, B.; McCormick, C. L. Prog. Polym. Sci. 2007, 32, 283.
(66) Georges, M. K.; Veregin, R. P. N.; Kazmaier, P. M.; Hamer, G. K. Macromolecules 1993, 26, 2987; Hawker, C. J.; Bosman, A. W.; Harth, E. Chem. Rev. 2001, 101, 3661.
(67) Golas, P. L.; Matyjaszewski, K. Chem. Soc. Rev. 2010, 39, 1338.
(68) Kolb, H. C.; Sharpless, K. B. Drug Discov. Today 2003, 8, 1128.
(69) Bock, V. D.; Hiemstra, H.; Maarseveen, J. H.-V. Eur. J. Org. Chem. 2006, 51, 2006.
(70) Rodionov, V. 0.; Fokin, V. V.; Finn. M. G. Angew. Chem., Mt. Ed. 2005, 44, 2210.
(71) Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Mt. Ed.
2002, 41, 2596.
(72) Himo, F.; Lovell, T.; Hilgraf, R. V.; Rostovtsev, V.; Noodleman, L.; Sharpless, K. B.;
Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210.
(73) Brase, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5518.
(74) Zhan, W. H.; Barnhill, H. N.; Sivakumar, K.; Tian, H.; Wang, Q. Tetrahedron Lett. 2005, 46, 1691.
(75) Mykhalichko, B. M.; Temkin, 0. N.; Mys'kiv, M. G. Chem. Rev. 2000, 69, 957.
(76) Chinchilla, R.; Najera, C. Chem. Rev. 2007, 107, 874.
(77) Horne, W. S.; Stout, C. D.; Ghadiri, M. R. J Am. Chem. Soc. 2003, 125, 9372.
(78) Hein, C. D.; Liu, X.-M.; Wang, D. Pharmaceutical Research 2008, 25, 2216.
(79) Nomura, K.; Geerts, Y. et al. Macromolecules 2008, 41, 4245; Macromolecules 2009, 42, 5104; Macromolecules 2011, 44, 3705.
(80) Nomura, K.; Miyamoto, Y.; Morimoto, H.; Geerts, Y. J Polym. Sci. PartA: Polym. Chem.
2005, 43, 6166.
(81) Nomura, K.; Abdellatif, M. M. Polymer 2010, 51, 1861.
(82) Fujiki, M.; Jalilah, A.; Suzuki, N.; Taguchi, M.; Zhang, W.; Abdellatif, M. M.; Nomura, K.
RSC Adv. 2012, 2, 6663.
Chapter 2
Precise synthesis of amphiphilic multi block copolymers by
combination of acyclic diene metathesis (ADMET) polymerization with atom transfer radical polymerization (ATRP), and click
chemistry
Contents
2-1.
2-2.
2-3.
2-4.
2-5.
Abstract Introduction Experimental
Results and Discussion
References and Notes
2-1. Abstract
Various block (graft) copolymers have been prepared by combination of acyclic diene metathesis (ADMET) polymerization of 9,9-dialkyl-2,7-divinyl-fluorene with Cu catalyzed atom transfer radical polymerization (ATRP) of styrene using macroinitiators prepared by introduction of initiating functionalities into poly(9,9-dialkylfluorene-2,7-vinylene)s (PFVs) chain ends: a precise synthesis of the amphiphilic ABCBA type block copolymers has also been attained by subsequent combination with click reaction after modification of the chain end with NaN3.
••• • ... (Br
O \.. ID 0
R R all-transn
Prepared by ADMET polymerization, end modification
ATRP i \
CuBr/dNbipy
;Grafting from DMF-THF NaN3
~Click Reaction Me0~~~0~/~O v \~
CuBr/dNbipy in THF
Grafting to
N /m
P-0
R R
ks-0-
O
d N
' Me()
O (
I' `N N •
O
0
—0 4:
i in
,mphiphlic ABCBA Block Copolymers 0
O n~
N-, N O O
OMe`
2-2. Introduction
Organic electronics are one of the most important emerging technologies, and conjugated polymers, such as poly(p-arylene vinylene)s, poly(thiophene)s, are promising semiconducting materials.1-4 Synthesis of structurally regular, chemically pure polymers by development of new synthetic methods attracts considerable attention,1 because their device performances are affected by polymer structural regularity, chemical purity, and supramolecular order33 Fluorene-based electroluminescent (EL) polymers are known to be promising in terms of a facile introduction of substituents into the C9 position, high photoluminescence (PL) and EL efficiencies, thermal and chemical stabilities.5-7
Nomura et al. recently demonstrated syntheses of defect-free, stereo-regular (all-trans), high molecular weight poly(9,9-dialkylfluorene-2,7-vinylene)s (PFVs), poly(2,5-dialkylphenylene-
1,4-vinylene)s (PPVs) by acyclic diene metathesis (ADMET) polymerization of 9,9-dialkyl-2,7-divinyl-fluorene.5'8-1° Since the resultant polymers prepared by Ru-carbene catalyst possessed well-defined polymer chain ends (as vinyl group),5b-d,9 a facile, exclusive end-functionalization can be achieved by treating the vinyl groups with Mo-alkylidene (Mo cat.) followed by Wittig-type cleavage with aldehyde:5b,d,11,12 Also precise syntheses of ABA type amphiphilic triblock copolymer by grafting PEG [poly(ethylene glycol)] into both the PFV chain ends,5b and of PFV's containing oligo(thiophene)s in the both chain ends which exhibiting unique emission properties by an energy transfer have been demonstrated.5d Formation of regular one-dimensional conjugated structures on the nanoscale should be thus expected by exploiting the specific assembling properties of rod-coil block copolymers,13,14 and the control of the block lengths via synthesis shall open the way to fine tuning the lateral dimensions of these nanostructures.
On the basis of the above background, in this chapter, various block (graft) copolymers have
been prepared by combination of ADMET technique with Cu catalyzed atom transfer radical
polymerization (ATRP) technique by using macroinitiators prepared by introductions of initiating
functionalities into PFV chain ends (grafting from approach).'5'16 Moreover, amphiphilic ABCBA
type block copolymers containing PEG fragment have been attained by additional combination
with click chemistry (grafting to approach).17
2-3. Experimental
Materials
General procedure
All experiments were carried out under a nitrogen atmosphere in a Vacuum Atmospheres drybox or using standard Schlenk techniques. All chemicals used were of reagent grades and were purified by the standard purification procedures. Anhydrous grade toluene (Kanto Kagaku Chemical Co., Inc.) was transferred into a bottle containing molecular sieves (mixture of 3A 1/16, 4A 1/8, and 13X 1/16) in the drybox, and was stored over sodium/potassium alloy in the drybox, and then passed through an alumina short column prior to use. Mo(CHCMe2Ph)(N-2,6- Me2C6H3)[OCMe(CF3)2]2 (Mo)18 was prepared according to the literature, and was used in the drybox. Polymerization grade of 2,7-divinyl-9,9-di-n-octylfluorene was prepared according to the previous report,19 and preparation of poly(9,9-di-n-octyl fluorene-2,7-vinylene) [Mn _ 1.96x104, M/Mri 1.94] by acyclic diene metathesis (ADMET) polymerization by ruthenium catalyst was according to the previous report.19b 4-(Bromomethyl)benzaldehyde, a-bromo-
isobutyryl bromide, sodium azide, 4-pentynoate terminated poly(ethylene glycol)methyl ether were also used in the dry-box as received (Aldrich Chemical Co.) without further purification.
All 1H and 13C NMR spectra were recorded on a Bruker AV500 spectrometer (500.13 MHz for 1H , 125.77 MHz for 13C) and all chemical shifts are given in ppm and are referenced to SiMe4.
Obvious multiplicities and routine coupling constants are usually not listed, and all spectra were
obtained in the solvent indicated at 25 °C unless otherwise noted. Molecular weights and the
molecular weight distributions of the resultant polymers were measured by gel-permeation
chromatography (GPC). HPLC grade THF was used for GPC and was degassed prior to use. GPC
were performed at 40 °C on a Shimadzu SCL-10A using a RID-10A detector (Shimadzu Co. Ltd.)
in THF (containing 0.03 wt % of 2,6-di-tert-butyl-p-cresol, flow rate 1.0 mL/min). GPC
columns (ShimPAC GPC-806, 804 and 802, 30 cm x 8.0 mm diameter, spherical porous gel made
of styrene/divinylbenzene copolymer, ranging from < 102 to 2x107 MW) were calibrated versus
polystyrene standard samples.
Synthesis of 2-bromo-2-methylpropionic acid 4-formyl-phenyl ester. Into a dichlorometathne (13 mL) solution containing a-bromoisobutyryl bromide (9.4 mL, 22 mmol) precooled to 0 °C, was added a dichlorometane solution (25 mL) containing p-hydroxy benzaldehyde (2.44 g, 20 mmol), triethyl amine (5.54 mL, 40 mmol) were added dropwisely over the period of 20 min under nitrogen atmosphere. The reaction mixture was allowed to warm to room temperature and stirred for 24 h. After the reaction the resultant white precipitate was filtered, and the filtrate was subsequently washed with 1M HC1, saturated aqueous Na2CO3 solution. The organic layer was dried over MgSO4, and was concentrated in vacuo. The resultant solid was recrystallized in the dry box using dry hexane, affording white microcrystals.
Yield 84.0 %. 1H NMR (CDC13 at 25 °C): 8 10.01 (s, 1H), 7.95 (d, 2H), 7.32 (d, 2H), 2.08 (s, 6H).
Synthesis of macroinitiator, PFV(C6H4CH2Br)2, PFV(C6H4OCOCMe2Br)2. In the drybox, a toluene solution (0.4 mL) containing poly(9,9-di-n-octylfluorene-2,7-vinylene) (10 mg, 0.82 limo', Mn = 1.23x10, MW/Mn = 1.94) was added Mo(CHCMe2Ph)(N-2,6-Me2C6H3)-[OCMe(CF3)2]2 (3 mg, 4.1 µmol, 5 equiv), and the solution was stirred for 3 h at room temperature. The mixture was then addede aldehyde [2-bromo-2-methylpropionic acid 4-formyl-phenyl ester, or 4-(bromomethyl)benzaldehyde] in excess (ca. >20 equiv) and was stirred 1 h for completion. The resultant polymer was precipitated by pouring the solution into methanol, and was collected by filtration. The precipitate was then dissolved in chloroform in the dry box, and was passed through alumina short column, and was dired in vacuo. Yield > 99.0 %.
2-Bromo-2-methylpropionic acid 4-formyl-phenyl ester as aldehyde for termination, PFV(C6H4OCOCMe2Br)2: 1H NMR (CDC13 at 25 °C): 8 7.69 (d, 2H), 7.53 (br, 4H), 7.22 (br, 2H, trans CH=CH-), 2.09 (s, 12H), 2.05 (br, 4H), 1.18-1.08 (br, 20H), 0.74 (t, 6H), 0.66 (br, 4H).
13C NMR (CDC1
3 at 25 °C): 8 14.1, 22.6, 23.7, 29.28, 29.7, 30.1, 30.67, 31.8, 40.6, 55.06, 119.9, 120.6, 121.3, 125.7, 127.3, 128.6, 136.5, 140.67, 151.6. PFV(C6H4OCOCMe2Br)2 M-(NMR) 12400 [estimated by integration ratio of aromatic protons vs - C(CH )2Br].
4-(Bromomethyl) benzaldehyde as aldehyde for termination, PFV(C6H4CH2Br)2: 1H NMR
(CDC13 at 25 °C): 8 7.69 (d, 2H), 7.53 (br, 4H), 7.22 (br, 2H, trans CH=CH-), 4.54 (S, 4H), 2.05
(br, 4H), 1.18-1.08 (br, 20H), 0.74 (t, 6H), 0.66 (br, 4H). 13C NMR (CDC13 at 25 °C): 8 14.1, 22.6,
23.7, 29.28, 29.7, 30.1, 31.8, 40.6, 55.06, 119.9, 120.6, 125.7, 126.8, 128.6, 129.5, 136.5, 140.67,
151.6. PFV(C6H4CH2Br)2 Mn(NR) = 12200 (estimated by integration ratio of aromatic protons vs -CH Br).
Atom transfer radical polymerization (ATRP): Synthesis of PFV-(PS-Br)2. A typical ATRP procedure is as follow. Into a sealed Schlenk tube, PFV(C6H4OCOCMe2Br)2 [Mn(NMR) _ 1.24x104] (10 mg, 0.81 µmol), CuBr (1.0 mg, 7 µmol, 8.5 equiv), dNbipy (4,4-dinonyl-2,2'- dipyridyl, 6 mg, 14µmol, 17 equiv), styrene (0.4 mL) were added in the drybox, and the tube was placed in an oil bath preheated at 90 °C. The mixture was stirred for prescribed time under dark conditions. The resultant polymer was precipitated by pouring into methanol, and was collected by filtration. The collected preciapitate was then dried in vacuo. (20 mg after 12 h, styrene conversion 5.0 %). 1H NMR (CDC13 at 25 °C): 8 7.69 (d, 2H), 7.53 (br, 4H), 7.22 (br, 2H, trans CH=CH-), 1.18-1.08 (br, 20H), 0.74 (t, 6H), 0.66 (br, 4H), and 7.16-6.82 (br, 3H), 6.81-6.23 (br, 2H), styrene phenyl ring, 2.05 (br, 3H), 1.63-1.32 (br, 3H, styrene aliphatic back bone). 13C NMR (CDC13 at 25 °C): 8 141, 22.6, 24.04, 29.28, 29.7, 30.1, 31.8, 40.6, 55.06, 119.9, 120.6, 125.7,
125.63, 136.5, 140.67, 151.6.
Click chemistry: Synthesis of PFV-(PS-N3)2. Into a dry vial, PFV-(PS-Br)2 [Mf(ivMR) 1.40x 104, Mn(GPc) = 2.86x10, MW/Mn = 1.70] (11 mg, 0.79 µmol), sodium azide (1 mg, 15.7µmol, 20 equiv), THF (1 mL) and dimethylformamide (1 mL) were added in the dry box and stirred overnight at room temperature, and the resulted polymer precipitated by pouring in methanol with stirring for 30 min, and was collected by filtration, The collected precipitate was then dried in vacuo, yellow precipitate is resulted. Yield > 99.0 %.
Synthesis of PFV-(PS-bI-PEG)2. Into a sealed Schlenk tube, PFV (PS-N3)2 [Mn(NMR)
1.25 x 104, Mn(Gpc) = 2.50x104, MW/Mn = 1.60] (5 mg, 0.41 µmol), 4-pentynoate terminated
poly(ethylene glycol) methyl ether [Mn = 2,000] (3 mg, 1.4 µmol, 3.5 equiv), CuBr (0.05 mg,
0.02 mmol, 5 equiv), dNbipy (8 mg, 0.04 mmol, 10 equiv) and THF (2 mL) were added in the dry
box, the mixture was stirred at 35 °C for 5 days. The resultant polymer was precipitated by
pouring in methanol, and was collected by filtration. The collected precipitate was then dried in
vacuo, yellow precipitate is resulted. Yield 90.0 %.1H NMR (CDC13 at 25 °C):8 7.69 (d, 2H), 7.53
(br, 4H), 7.22 (br, 2H, trans -CH=CH-), [7.16-6.82 (br, 3H), 6.81-6.23 (br, 2H) styrene phenyl
ring], 3.62 (br), 2.05 (br, 4H), 1.63-1.32 (br, 3H, styrene aliphatic back bone), 1.18-1.08 (br,
20H), 0.74 (t, 6H), 0.66 (br, 4H). 13C NMR (CDC13 at 25 °C): 8 10.9, 14.06, 14.13, 14.19, 22.6,
22.7,22.99,23.7,26.7,28.9,29.2,29.7,30.1,30.3,31.8,31.9,33.7,38.7,40.7,55.04,68.1,
70.58,119.9,120.6,125.7,128.6,128.8,130.8,132.4,136.5,151.5,167.7.
2-4. Results and Discussion
Acyclic diene metathesis (ADMET) polymerization of 2,7-divinyl-9,9-di-n-octylfluorene was conducted in the presence of Ru catalyst according to the established conditions under a reduced pressure (reaction time 3 h, Scheme 2_1) 5b-d The resultant poly(9,9-di-n-octylfluorene- 2,7-vinylene) (PFV) possessed high molecular weight with unimodal molecular weight distribution (by GPC analysis: Mn = 1.96x 104, MwlMn = 1.94). As demonstrated previously,5 the resultant PFVs possessed exclusive trans regularity as well as vinyl groups at the both polymer chain ends as confirmed by 1H NMR spectra [S 7.22 (br, 2H, trans -CH=CH-) and coressponding peaks for terminal vinl group 6 6.74 (dd, 2H), 6.74 (dd, 2H), 6.74 (dd, 2H), (Figure 2-la)I.51-d
According to the reported procedure shown in (Scheme 2-1),51-d the vinyl groups at the PFV
chain ends were treated with Mo cat. (2.5 equiv to the vinyl group, to generate Mo-alkylidene
moieties) and the subsequent addition of excess amount (ca. 2 equiv to Mo) of various aldehydes
(ArCHO) gave PFVs containing functionalities at the both polymer chain ends. Protons
corresponding to the terminal vinyl disappeared and the corresponding one of chain ends could be
observed in the 1H NMR spectra (Figure 2-lb,c) [8 2.09 (s, 12H, —C(CH3)2Br) in case of
PFV(C6H4OCOC(CH3)2Br)2 and 5 4.54 (S, 4H, -CHBr) in case of PFV(C6H4CH2Br)], and The
Mn values for PFV(C6H4CH2Br)2, PFV(C6H4OCOCMe2Br)2 estimated by 1H NMR spectra (on
the basis of aromatic protons in PFV vs initiating fragment) were 1.22x 104, 1.24x 104,
respectively. These values are relatively close to the estimated values (Mn = 1.25 x 104, 1.27x 104)
on the basis of the exact value of PFV estimated by GPC [Mn(corrected) = 1.22x 104, Mn(GPC) _
1.96x 104] and the initiating fragment. As reported previously,5a Mn value in GPC vs polystyrene
standards is higher than that in GPC vs PPP standards, because of the nature of rigid conjugated
polymers, and the estimation [Mn(calcd.) = Mn(Gpc)/1.6] was adopted for further study,5 strongly
suggesting that both polymer chain ends could be exclusively modified by adopting the present
approach.21
n
R R
• R = n-octyl, C8H17
ArCHO
OHC * OHC *
O C----Br O
CH2Br
Ru cat. =Ar—N N—Ar
in tolueneADMET Polymerization
\luCIi, RuCHPh CH2=CHOEt1~CI
(termination)Ru cat. PCy3
Ar = 2,4,6-Me3C6H2,
R RCy = cyclohexyl
PFV n-1••\'.. II I
i) Mo cat.
(5 equiv.) ii) ArCHO
(F3C3)2
(F3C)MeCO`~Nt 2MeCO°'CHCMe2Ph Mo cat.
Br
o * pi... --
PFV(C6H4O0OCMe2Br)2
BrH2C ^'•
PFV(C6H4CH2Br)2
n-1\\
n-1 R
R R
•
O
--- Br
0
CH2Br
Scheme 2-1. Syntheses of macroinitiators [PFV(C6H4CH2Br)2, PFV(C6H4000CMe2Br)2]
33
(a)
e f
CDCI3
PFV
Mn=1.96x104, MW/Mn=1.94
x.
-CH=CH2
is ca s'5
C H2O din CDCI
a
b
Br-
Br - 1/
iR R
_x
R=n-octyl
RR t.131
PFV(C6H4CH2Br)2 H
.,x(b) CDCI3 H20inCC 3
x
ai I 1.
Ail 4 'S3 ppm
SiMe4 7/
R=n-octyl
RR
R R
O
y ~.
-FV( 6H4000CMe2Br)2 (c)
H2O in CDC!,
Me CDCI3
7.5 7.0 55 6.0 5.5 5.0 4.5 4.0 3.5 10 2.5 2.0 1.5 1.0 0.5 ppm
Br
Figure 2-1. 1 H NMR spectra of (a) PFV, (b) PFV(C6H4CH2Br)
PFV(C6H4OCOCMe2Br)2
2 and (c)
34
The resultant macroinitiators, PFV(C6H4OCOCMe2Br)2, PFV(C6H4CH2Br)2, were added in styrene in the presence of CuBr, dNbipy (4,4-dinonyl-2,2'-dipyridyl) at 90 °C for conducting subsequent atom transfer radical polymerization (ATRP, Scheme 2-2) .
Br—IM 40 \ 41,--11R R
R R n-1 •--O \
IM = OCOMe2C or CH2 C
uBr/dNbipy
Initiating moiety° /90C
M®\\R R
BC m441\
®R R n-1
poly(styrene-b/-PFV-b/-styrene)s PFV-(PS-Br)2
44I IM-Br
le -Br
//
Scheme 2-2. Syntheses of PFV-(PS-Br)2 by Cu catalyzed ATRP of styrene initiated by PFV(C6H4OCOCMe2Br)2, PFV(C6H4CH2Br)2
The NMR spectra of the resultant copolymers (Figure 2-2, Figure 2-3), indicate that styrene repeat units were incorporated and the styrene contents [in the 1H NMR spectra aromatic protons of styrene b 7.16-6.82 (br, 3H), 6.81-6.23 (br, 2H) and styrene aliphatic back bone 6 2.05 (br, 3H),
1.63-1.32 (br, 3H)], increased over time course (upon increasing
+ the yields, conversion). As shown in (Figure 2-4), relatively linear relationships between Mn values estimated by their 1H NMR spectra (based on integration ratios of protons between styrene and PFV) and the conversion or polymerization time were observed, and relatively close relationships were observed when the M„ values by GPC were employed for the plots. These would suggest that these polymerizations proceeded in a living manner, the results are summarized in (Table 2-1).
35
Br 0
0
i0-01\((v.
R=n-octyl
run 5, Table 1
\ /„
rtI®
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3,5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
Figure 2-2. 1H NMR spectra of PFV-(PS-Br)2 initiated by PFV(C6H4OCOCMe2Br)2
Br 0
0
Ia
L,




![Table 2-1. Synthesis of block copolymers, poly(styrene-bl-PFV-bl-styrene)s [PFV-(PS-Br)2], by Cu catalyzed atom transfer radical polymerization (ATRP) of styrene initiated from macroinitiators containing PFV [PFV(C6H4CH2Br)2 or PF](https://thumb-ap.123doks.com/thumbv2/123deta/10115837.1949816/41.850.61.789.43.1225/synthesis-copolymers-catalyzed-transfer-polymerization-initiated-macroinitiators-containing.webp)
![Table 3-1. Synthesis of oligo(2,5-dialkoxy-1,4-phenylene end functional groups [alkoxy (OR) = O(CH2)2OSi'Pr3].°](https://thumb-ap.123doks.com/thumbv2/123deta/10115837.1949816/63.850.100.754.248.626/table-synthesis-oligo-dialkoxy-phenylene-functional-groups-alkoxy.webp)