Vol .
15
No.
2
July 2014
ISSN 2187-798X目 次
1
リレーエッセイ〈6〉「次世代」がやってきた
田村浩一郎
(首都大学東京・生命情報研究センター、日本進化学会副会長)3
平成26年度科学技術分野・文部科学大臣表彰受賞者のお知らせ
4
第16回日本進化学会大会(大阪大会)のご案内
5
進化学者に聞く! 学生からの10の質問
入江直樹
(東京大学大学院 理学系研究科)長田直樹
(国立遺伝学研究所、総合研究大学院大学)8
ミーティングレポートエボ・グループセミナー in コーネル大学
山道真人
(京都大学・白眉センター)9
新シリーズ「大量データと知見の架け橋」(
緒言・編集担当佐藤行人)
第1回「RNA-Seqによる網羅的トランスクリプトーム解析」
成相直樹
(東北大学 東北メディカル・メガバンク機構 ゲノム解析部門)16
海外研究室だより 第18回ジュネーブからケンブリッジへ:エピジェネティクス研究室紹介
岩崎まゆみ
(ケンブリッジ大学、Sainsbury Laboratory)19
書 評「ヒトは病気とともに進化した」
「クワガタムシが語る生物多様性」
「From Taxonomy to Phylogenetics:
Life and Work of Willi Hennig
」
荒木仁志
(北海道大学大学院・農学研究院)三中信宏
(農業環境技術研究所・生態系計測研究領域)24
編集後記
日本進化学会ニュース July 2014 1 リレーエッセイ ︿ 6﹀ ﹁次世代﹂ がやってきた 大学に勤めて20年にもなると、その間いろいろな変化を感じるものだ。中でもとりわけ大きな変化が「次 世代」の到来だ。いわゆるジェネレーションギャップというものか。大学に助手(助教)として赴任した当時、 学生、特に研究室に所属する大学院生は、教員と学生という立場の違いはあるにせよ、一言で言えば「後輩」 だった。それが今では自分の子供と同年代になってしまい、「後輩」から「子供」になってしまった。学生の方 から見れば「親と同じ歳」ということになり、こちらがいくら「先輩」のつもりでいても、それは許してはもら えず「親」と同属の扱いを受けてしまう。まあ、子供に甘い親然り、若いころには違和感を覚えた、教授の学 生に対する優しすぎる態度が理解できるようになったのは収穫かもしれない。今はさぞかし若手から違和感 を持たれていることだろう。 さて、この進化ニュースで書きたかったのは、オヤジの心境の変化ではもちろんない。「次世代」とは次世 代学生のことではなく、次世代DNAシーケンサー、NGS(Next Generation Sequencer)のことだ。生命科 学のどの分野でもそうかもしれないが、進化研究分野では、その恩恵はとりわけ大きいのではないかと思う。 長谷部さんが、リレーエッセイ〈4〉の最後で「その気になればどんな材料でも研究できる時代になったので、 今後、いろいろ生物の研究がさらに進む…」と言ったのはまさにこのことだ。いろいろな種について比較解析 が必要な進化研究では、モデル生物のみに注目していればいい分野とは違った難しさがあった。例えば、興 味ある非モデル生物種についてマイクロアレイを活用しようとすれば、独自にチップを作らなければならな かった。それがRNA-seqならば、特別な準備をすることなく網羅的発現解析ができる。NGSのおかげで、近 縁種間で発現が異なる遺伝子を見つけることなど今や容易いことになったのだ。すでにメジャーなジャーナル では、NGSを使って種間で発現パターンの異なる遺伝子を見つけただけでは、論文としてアクセプトしても らえない状況になった。ちょっと前までなら夢のような話だったのだが、技術の進歩とは恐ろしいものだ。そ の後に続く研究が楽しみになったことは大歓迎なのだが。いずれにしろ、NGSがやってきたことで、研究の やり方が大きく変わった。 我々の研究室に最初にNGSが来たのは、約3年半前のことだ。大学の老朽備品更新の機会に、ジャンク同 然だったABI310をRoche 454 GS Jrに買い替えてもらった。いい買い物だったかどうかは今となっては怪し いが、当時はそのような価格帯で買える機種が登場したことが画期的で、他に選択の余地はなかった。一回 のランで平均450 bpの断片が10万リード得られる能力があり、そのために必要な試薬キットの価格は十数万 円である。高いのか安いのかは分からないが、特別な会計手続き無しに購入できる価格は魅力的で、少なく ともこれでパイロット実験を行い、見通しが立てば専門業者への外部委託で本格的なデータを得るというの がベストな使い方のように思われた。このNGSの到来によって、学生の間でも意識の変化が生じたような気 がする。 NGSを使うために重要なことは、基本的な実験操作が確実で精度が高いことだ。一般に、DNAを扱う分 子生物学的実験法は定性的で、厳密な定量性は必要ない。例えば、核酸のアガロース電気泳動では、数十 ngのDNAを用いればバンドを程よく見ることができるが、それが数ngになってもぎりぎり見えるし、数百 ngでも問題ない。PCRに至っては、鋳型DNAが入っているかいないかの差が増えるか増えないかの結果に つながるようなものだ。一言で言えば、物の量は桁が合っていれば大抵うまくいく。ところが454 GS Jrシー ケンサーのための反応では、50万粒のエマルジョンPCR用ビーズにかなり正確に11万分子のDNAを加える 必要がある。この条件だと平均して5粒中1粒のビーズにDNAが付着する。1粒のビーズに複数のDNAが 付着する確率は5%程度なので、その分と+αを差し引いて約10万リードが得られる算段だ。加えるDNAの
リレーエッセイ
〈
6
〉
「次世代」がやってきた
田村浩一郎
(日本進化学会副会長)July 2014 2 リレーエッセイ ︿ 6﹀ ﹁次世代﹂ がやってきた 量が少なければリード数はその分少なくなるが、多くても複数のDNA分子が付着するビーズが増え、その分 リード数は減る。つまり、最適な分子量のDNAを最適な量加えなければ、良好なリード数は得られないしく みになっている。このようなNGSの特性を知ると、NGSを使いたいがために、学生の実験技術向上のモチ ベーションは上がる(ような気がする)。実験用の装置、器具、試薬キットの発展によって、誰でも簡単にで きる分子生物学的実験は少々退屈だが、NGSの使用はいい刺激になっていると思う。 しかし、それ以上の変化は、コンピュータ・プログラミングへのモチベーションが格段に上がったことだ。 研究の重心がwetからdryへ移動したと言ってもいい。出力される10万リードの配列データを、手作業で処 理するのはさすがに辛い。これまでは「簡単なプログラムを作ればすぐできる」と嗾けても「手動で頑張った 方が早い」と答えていた学生が、今度は「手動でやるのは絶対にイヤ、プログラムを作ります」と言い出した。 アセンブルやマッピングなど、基本的なことは既存のプログラムを利用すればいいが、BLASTを活用して オーソログを判別し、それらの配列アライメントを作るというような、一歩進んだ独自の解析をやるには、あ る程度のプログラミング技術が必要になる。NGSを使って生データを出すまでのwetな作業より、それを進 化研究に活用できるようにするまでのdryな作業の方が、明らかに手間暇がかかるようになった。今後さらに wetな実験のキット化が進む一方、NGSの出力データ量、ランニングコストが改善されれば、この傾向はさら に進むだろう。dryな作業の腕前が直接、研究成果の良し悪しに結びつくようになるのではないか。グローバ ル化に伴い英語能力の重要性が声高に議論されるようになったが、次世代化に伴いバイオインフォ能力の重 要性はそれ以上に高くなっている。今の学生や若い研究者の皆さんには、是非、コンピュータの勉強にも力 を注いでほしいと思う。 Roche 454 GS Jrシーケンサーは、発売開始して3年余りで生産中止、消耗品のサポートも今後3年間で終 了することになった。「いい買い物だったかどうかは怪しい」とはこのことだ。ハイテク機器の命は儚く、後 進のIllumina MiSeqの追従に耐えかねたのだろう。最新の試薬キットを用いれば、1回のランで300 bp×2 (300 bpのペアエンド=500 bp)の断片が2,500万リード得られる。機器の価格は似たり寄ったりで、試薬キッ トの価格も倍はしない。それでいて200倍以上のデータが得られるのだから、これはもう勝負にならない。コ スパが100倍違う。Illuminaの利点はデータの質にもある。454の場合、原理的に「AAAA」のように同じ塩 基が続くホモポリマーの解析に難があり、連続した塩基の数の読み違えが高頻度で起こる。その結果、アセ ンブルを行う際にはリード間で局所的にアライメントを行う必要があり、そこにかなりの計算時間を要する。 Illuminaはこのような問題が無いため、桁違いのリード数でもアセンブルを高速で行える。代わりに塩基置 換型のシーケンシングエラー(リードミス)は454よりも多く起きるようだが、リード数の多さでカバーしても 十分おつりがくる。この春、我々の研究室でもIllumina MiSeqを手に入れ、454 GS Jrが置いてあった場所 に設置した。これから活用しようというところである。哀れな454 GS Jrではあるが、一足早くNGSを経験で きたことはよかったと思っている。 Illuminaの導入によって、研究室のdry環境の整備、充実化がさらに進むことになった。数千万リードの 配列をde novoアセンブルするためには、メモリ量とコア数の充実した高性能コンピュータが必要になる。特 に既存のソフトを活用するためには、Linuxマシンの使用が欠かせない。現在、一番よく使っているのは、
DELL T3600(Xeon 6コア×2、64 GB RAM)ワークステーションで、OSはUbuntu ver. 12.04(Linux)をイ
ンストールしている。外注で出した75 bp、3,500万リードのcDNAを、Velvetを使ってde novoアセンブルし てみたところ10分くらいでできてしまったが、2セットのデータを合わせて7,000万リードにするとメモリが 足りなくなった。最近、たまたま中古のサーバをタダ同然で手に入れたので、メモリを192 GBに増設して新 たな解析マシンに仕立て上げようと計画している。今やバイオインフォマティシャンというよりは、ほとんど SEになってしまった。学生にとっては、コンピュータのハードウェアをいじるよりプログラミングの方がまだ 馴染みやすいようだが、今後はハードウェアの知識もあれば得するのは確実だ。例えば、中古市場でジャン クマシンを買ってパーツを買い足せば、新品で買えば数百万円するコンピュータもその十分の一の価格で構 築することができる。NGSが高性能・低価格化すると、必然的に課題が解析用のコンピュータに移っていく
日本進化学会ニュース July 2014 3 平成 26年度科学技術分野・文部科学大臣表彰受賞者のお知らせ
平成
26
年度
科学技術分野・文部科学大臣表彰受賞者のお知らせ
日本進化学会から推薦した中野裕昭さん(筑波大学 下田臨海実験センター)が、文部科学大臣表彰若手 科学者賞を受賞されましたので、お知らせいたします。 詳しくはこちらをご覧ください。 http://www.mext.go.jp/b_menu/houdou/26/04/1346090.htm http://www.mext.go.jp/b_menu/houdou/26/04/__icsFiles/afieldfile/2014/04/07/1346090_02.pdf なお、他団体からの推薦ですが、中野さん以外にも以下の3名の会員が若手科学者賞を受賞されています。 二橋 亮さん(産総研 生物プロセス研究部門) 細 将貴さん(京都大学 白眉センター) 舞木昭彦さん(島根大学 生物資源科学部) 受賞者の皆様、おめでとうございます。 と予想される。コンピュータの知識は、知っていて損はないどころか、かなり得することになるだろう。 MEGAの開発を手掛けてから20年が過ぎた。今、20年前の自分だったら、きっとNGS関連の解析ソフトウェア開発に手をだし、「GIGA(Genome Informatics and Genetic Analysis?)」とか作っているのだろう、 と想像したりする。新しいテーマにむけて腕を磨くには、今がチャンスだと思うのだが、いかがだろう。 著者紹介 田村浩一郎 所属:首都大学東京 生命情報研究センター/院・理工・生命科学専攻 最終学歴:理学博士・1991年(東京都立大学理学研究科生物学専攻) 職歴(略歴): 1991年4月∼ 1993年3月 ペンシルバニア州立大学 分子進化遺伝学研究所 ポスドク研究員 1993年4月∼ 2005年3月 東京都立大学 理学部 助手 2005年4月∼ 2008年9月 首都大学東京 理工学研究科 准教授 2008年10月∼現在 首都大学東京 理工学研究科 教授 2013年1月∼現在 生命情報研究センター センター長
July 2014 4 第 16回日本進化学会大会 ︵大阪大会︶ のご案内 日程 2014年8月21日(木)∼ 24日(日) 会場 高槻現代劇場(阪急高槻市駅より徒歩5分、JR高槻駅より徒歩12分) 大会ホームページ https://sites.google.com/site/shinka2014osaka/ 第16回日本進化学会大阪大会を、2014年8月21日(木)午後から24日(日)の4日間、大阪府高槻市の現 代劇場で開催します。22日午後には、国際プレナリーシンポジウムを大阪大学の四方哲也博士に企画して頂 いています。「実験室内進化」をメインテーマとし、海外からSantiago F. Elena博士(サンタフェ研究所、ス ペイン分子細胞生物学研究所)、Tim Cooper博士(ヒューストン大学)を招き、最新の研究動向を紹介して 頂きます。 21日∼ 24日の全期間においてシンポジウムやワークショップが行われます。その内容は、激論、生命の起 源:海か陸か?初期地球での生命誕生プロセス、顕生代の再定義と再評価、環境変化に適応した形態進化、 節足動物の多様性と進化、シーラカンス研究の最前線、極限環境適応、適応進化の生態的帰結、進化精神医 学の幕開け、現生人類起源論、宿主・ウイルス共進化、エビゲノム進化、コドンの誕生と進化、突然変異メ カニズム、多面的な進化学研究、次世代シークエンスを活用した進化解析など、多岐にわたります。また一 般演題(口頭とポスター)も並行して行われます。すべてのポスターは初日から3日目午後まで掲示できます (ポスター会場は懇親会会場に模様替えします)。 3日目23日の午後には、高校生ポスター発表「第9回みんなのジュニア進化学」と、一般市民向けに公開講 座「生きものはつながりの中に」を開きます。公開講座では、生命誌研究館20周年を記念して制作した演劇 「生命誌版 セロ弾きのゴーシュ」の上演と「共生で進化する生命」と題した深津武馬博士の講演が行われま す。この公開講座は入場無料ですが、入場には整理券が必要です。学会会員向けには当日券を用意していま す。また、同日10時から16時まで、学会会員による研究紹介企画「進化って何だろう? 研究者と話してみ よう!」を行います。芸術とサイエンスの融合、一般市民と学会員の交流を意図した企画です。 最終日24日午後の進化学・夏の学校ではNGSデータ解析デモンストレーションを開催します。RNA-seq などNGSデータを解析する際の、具体的な手順やコマンドを最前線で活躍する研究者が紹介する予定です。 多くの皆様のご参加をお待ちしております。 <大会概要> 8月21日(木) 一般口頭発表、シンポジウム、ワークショップ、ポスター掲示 8月22日(金) 一般口頭発表、シンポジウム、ワークショップ、ポスター発表、プレナリー国際シンポジウム 8月23日(土) 午前:ポスター発表、ワークショップ、研究紹介展示 午後:高校生ポスター発表、公開講座(入場無料)、研究紹介展示、高校生ポスター表彰式、 総会・学会賞授賞式・受賞講演、懇親会 8月24日(日) シンポジウム、ワークショップ、進化学・夏の学校(入場無料) 最新情報については随時大会ホームページ(https://sites.google.com/site/shinka2014osaka/)をご確認く ださい。 大会委員長:蘇智慧(JT生命誌研究館) 大会準備委員長:小田広樹(JT生命誌研究館) 大会事務局メールアドレス:[email protected]
第
16
回日本進化学会大会(大阪大会)の
ご案内
日本進化学会ニュース July 2014 5 進化学者に聞く! 学生からの 10の質問 「進化に興味はあるものの、周りには何をやっているのか分かってもらえないし、将来が不安…」と感じて いる読者も多いのではないでしょうか。そこで、実際に進化学研究で世界をリードする日本進化学会執行部 の先輩方に、あれこれ聞いてみることにしました。今回は会計幹事の入江直樹さん、庶務幹事の長田直樹さ んのお二人です。 まずは入江さんの答えから。
Q1
進化学者になったきっかけを教えてください。A1
動物発生システムの普遍的性質を探求したいと思ううちに行き着きました。Q2
進化学者になってよかった、と思った瞬間はいつですか?A2
UK・ケンブリッジに出張中、布教活動をしている仏教徒に興味深そうにいろんな質問をされた時。 Shintoなのか?進化生物学者としてどのような人生を考えているのか。あなたにとって人生は整合的か。Q3
小さい頃の夢を教えてください。A3
大工→パイロット→宇宙人来襲時に地球防衛軍に呼ばれるような科学者になりたい。Q4
研究者になるのを諦めかけたこと、ありますか? もしあればどうやって克服を?A4
大学院生の間に自分の着想で研究をスタートさせられなかったら、科学者になるのを諦めるつもりでし た。…が、どうにか自分の着想ではじめた小さな研究で学位を取得。Q5
進化学者になるのに必要な素質・スキルって何でしょう?A5
「これ、数億年前のあなたの祖先の姿かもしれません」→「おぉ∼」って感動できる力、、でしょうか?Q6
学部生・院生当時の一番の思い出は?A6
京都に訪れていた免疫分野のノーベル賞受賞学者(Dr. Rolf Martin Zinkernagel)の講義を聴いた時。質疑応答時の現役の免疫学者の先進性にむしろ衝撃を受けた(免疫系の司令塔やがん免疫についての 話など)。
Q7
現在の研究内容について教えてください。A7
動物のからだのうち、つくりかえにくい(発生学的)仕組みというのは存在するのだろうか?脊椎動物 亜門はなぜ脊椎動物門ではないのか。門や亜門などの高次分類群の議論自体不毛なのだろうか。脊索 動物のなかで、共通祖先から一番特殊化した動物はどれ?ということが言えるのだろうか?などなど。Q8
今後の研究展開や抱負を聞かせてください。A8
進化発生学という分野がありますが、マクスウェルの4方程式のように電場と磁場の両方を説明するよ うな、進化と発生の両方を統一的に説明する完成度の高い理論はないです。進化の予測性についても 語れる日がきてほしい。そういった問題に挑戦したい。進化学者に聞く!
学生からの
10
の質問
?
入江直樹
(東京大学大学院 理学系研究科)長田直樹
(国立遺伝学研究所、総合研究大学院大学)July 2014 6 進化学者に聞く! 学生からの 10の質問
Q9
10年後、進化学はどこまで進んでいると思いますか?A9
発生システムの進化スケールでの可変性について、もう少し議論が進んで、、いて欲しい。Q10
未来の進化学者に一言。A10
新しい世界観をつくるのは、大御所先生達ではなくみなさんです。 研究者紹介 入江直樹 所属:東京大学 院・理・生物科学専攻 最終学歴:博士(医学)・京都大学大学院 医学研究科 分子医学系専攻・2008年 職歴(略歴): 2008年4月∼ 2009年3月 京都大学 先天異常標本解析センター ポスドク研究員 2009年4月∼ 2013年8月 理化学研究所 発生・再生科学総合研究センター 研究員 2013年8月∼現在 東京大学大学院 理学系研究科 准教授 次は、長田さんです。Q1
進化学者になったきっかけを教えてください。A1
自分の興味の赴くままに研究をしていったら、気づいた時には進化の世界に入っていました。学位をと るまでは進化よりもゲノム科学が専門でしたが、その過程で進化について勉強することの重要性に気付 きました。学位取得後、進化研究で有名なシカゴ大学にポスドクとして行く機会に運よく恵まれ、より 深く進化学にコミットすることができたと思います。Q2
進化学者になってよかった、と思った瞬間はいつですか?A2
正直に言うと、なってよかったと思った瞬間はありません。ただ、これまでを振り返ってみると、好き なことができて楽しい研究生活だったと思います。Q3
小さい頃の夢を教えてください。A3
あまり将来についてまじめに考えたことはありませんでしたが、ほかの人とは何かしら違ったことをや りたいと思っていました。小さい時はご多分に漏れず昆虫少年でした。子供の頃は生き物を飼うのが好 きでしたが、最近は世話が上手くできないので生き物は飼わないようにしています。Q4
研究者になるのを諦めかけたこと、ありますか? もしあればどうやって克服を?A4
実のところ、学生の時はあまり研究に身が入りませんでした。ポスドクの職を得て、研究で飯を食って いるのだと自覚してからやる気が出てきた記憶があります。気が付いたら惰性で研究をしていないか定 期的に自己点検をし、研究のモチベーションを高めることは必要だと思います。Q5
進化学者になるのに必要な素質・スキルって何でしょう?A5
進化学者に限りませんが、好奇心と論理的な思考力は研究者にとって重要だと思います。前者があれ ば研究生活が楽しくなりますし、後者があれば時間を無駄にすることが少なくなります。また、進化学 は歴史の長い学問なので、過去の研究によく学ぶことがとても大切だと思います。Q6
学部生・院生当時の一番の思い出は?A6
僕は人類学を専攻していましたので、色々な種類の実習に参加をしたのが記憶に残っています。北海日本進化学会ニュース July 2014 7 進化学者に聞く! 学生からの 10の質問 道に遺跡発掘(という名の土木作業)に行ったり、長野にサルを観察しに行ったり、自分の遺伝的体質 を知るために乳糖をとってみたり、山形に石器を作りに行ったりと、なんだかよくわからない実習をい ろいろと行いました。
Q7
現在の研究内容について教えてください。A7
生物の進化メカニズムについて、遺伝情報(特にゲノム情報)を用いて研究しています。特に、様々な 進化理論を現在の膨大な遺伝子データを用いてどのように証明できるかについて考えています。最近は 次世代シークエンサーの普及によりいろいろな生物の塩基配列データが大量に手に入るようになりまし た。自分で興味のある生物のゲノムデータを取得することと、すでに公になっているデータの再解析を することの二本立てで研究を進めています。Q8
今後の研究展開や抱負を聞かせてください。A8
僕は研究対象となる生物種があまり決まっていないので、動物でも植物でも興味があればなんでも解析 します。あまり複雑なことはやりたくないのですが、最近は遺伝子の相互作用、特に自然選択が比較的 弱い時の相互作用に注目した進化の研究を進めています。Q9
10年後、進化学はどこまで進んでいると思いますか?A9
10年後に可能かどうかはわかりませんが、生物のゲノム配列の解析コストが驚くほど安くなれば、研究 対象のゲノムをすべて解析するということが当たり前になってくると思います。とってきたサンプルの ゲノムが全部わかる時代になれば、その集団がどのような歴史を経て進化してきたのか、遺伝子型と表 現型がどのようにかかわっているのかがより詳しくわかるようになります。そのためには誰でも使える 簡単な集団ゲノム解析ツールが必要になるでしょう。もう一点は、脳科学分野の進歩により、行動など の複雑な形質の遺伝基盤がわかるようになるかもしれません。行動などの非常に複雑な形質がどのよう に進化してきたのかというのは進化学の究極の問いの一つです。ある程度高等な生物において、ゲノム 配列からある環境における行動を高い精度で予測できるようになれば、その行動がどのように進化して きたのかを知ることができるようになるかもしれません。そのためには遺伝子型からどのように表現型 を予測するかについての研究が大切になってくると思います。Q10
未来の進化学者に一言。A10
進化研究はますます多様化してきており、これから進化の研究を始める若い研究者は、情報の渦に飲み 込まれて何をしたらよいのか見失ってしまうかもしれません。そういった時には最先端の知識を追い求 めるだけでなく、過去の研究の歴史を振り返ってゆっくり物事を考えてみることも重要だと思います。 研究者紹介 名前:長田直樹 所属:国立遺伝学研究所 進化遺伝部門 最終学歴:博士(理学)、東京大学大学院生命科学専攻、2002年 職歴(略歴): 2002 東京大学 博士研究員 2003 ∼ 2004 シカゴ大学 リサーチアソシエイト 2004 ∼ 2005 国立感染症研究所 研究官 2005 ∼ 2009 独立行政法人医薬基盤研究所 研究員 2010 ∼現在 国立遺伝学研究所・総合研究大学院大学 助教July 2014 8 ミーティングレポート エボ・グループセミナー inコーネル大学 私は2012年4月から2014年3月にかけて、日本学術振興会の海外特別研究員として米国ニューヨーク州イ サカのコーネル大学で研究を行っていた。イサカはマンハッタンから車で5時間ほど離れた小さな街だが、ア イビーリーグの一翼を担う巨大なコーネル大学を擁するため、進化生物学の研究者も数多く住んでいる。本 稿では、コーネル大学における進化生物学研究者の交流の様子についてご紹介したい。
コーネル大学では、進化生物学者は多様な学科に分かれて所属している。私が籍を置いていたDepart-ment of Ecology and Evolutionary Biology(EEB)には非モデル生物の適応・種分化・系統地理の研究者
が、隣のNeurobiology and Behaviorには神経行動学者がおり、理論研究者の所属は両者とApplied
Math-ematicsに及ぶ。ショウジョウバエの集団遺伝学者はDepartment of Molecular Biology and Geneticsと Entomology(昆虫学)に分かれているし、Biological Statistics and Computational BiologyやMicrobiology、 Plant Biologyにも進化的な視点から研究を行う人々がいる。このような進化生物学者同士の交流を深めるた め、EEBの大学院生が中心になって、月1回のセミナーと年1回のシンポジウムが開催されている。 セミナーはEvo-group seminarと呼ばれ、月の最初の木曜日の夕方に1時間かけて行われる。講演は30分 ずつ、2人の演者によって行われ、基本的に大学院生やコーネル大学に移ってきたばかりのポスドクが発表す ることが多い。セミナーの後は大学内のホテル(Statler Hotelと呼ばれ、ホテルスクールの学生によって運営 されている)のバーに移動し、ビールを飲みながら更に議論が続けられる。 シンポジウムは毎年春にコーネル大学のLab of Ornithology(鳥類学研究所)において開催される。鳥類 学研究所はイサカの街の中心部から離れているため、普段訪れる機会が少ない場所である。池と森に囲まれ 遊歩道も完備された、バードウォッチングに最適な研究所で、鳥類学の研究拠点として全米に名を馳せてい る(図
1
)。進化生物学のシンポジウムは毎年異なるテーマで開催されており、2012年は種分化(Speciationエボ・グループセミナー
in
コーネル大学
山道真人
(京都大学・白眉センター)ミーティングレポート
図1 シンポジウムが開催されるコーネル鳥類学研究所(正式には、鳥類学研究 所の本部があるImogene Powers Johnson Center for Birds and Biodiversity)。 左手奥の塀の 間から鳥を観察することができる。日本進化学会ニュース
July 2014
9
新シリーズ
﹁大量データと知見の架け橋﹂
Day)、2013年は新奇形質と迅速な進化(Evo-day 2013: Novel Traits and Rapid Evolution)、2014年は性選
択(Evo-day 2014: Sexual Selection)であった。発表者はコーネル大学だけでなく、イサカから車で1時間の シラキュース大学、ビンガムトン大学、3時間ほどかかるロチェスター大学といったニューヨーク州内の大学 からも招待され、朝から夕方まで講演を行う。まれにハーバード大学やシカゴ大学といったやや遠い大学の 研究者も発表するが、米国北東部に限定されているようだ。朝食と昼食の時間には、廊下で院生・ポスドク のポスター発表も行われる。講演終了後はビールを飲みながら夜まで議論が続けられ、1日中そのテーマにつ いて語り尽くすことができる。 これらのイベントは基本的に大学院生によって運営されている。院生にとっては、自分の興味のある研究 テーマについて多様な学科の研究者と議論を深められる他に、有名な研究者にコンタクトを取りシンポジウム に招待する、会場と食事を確保し多くの人に向けて宣伝するといった一連の作業を経験できる点でも、得難 いイベントになっていると思われる。 私は2012年の種分化シンポではポスター発表を行い、秋にセミナーで発表し、2013年の新奇形質・迅速 な進化シンポでは口頭発表を行った。これらは、それまでこちらが一方的に知っていた進化生物学の教授た ちと知り合う機会になるのみならず、同年代のポスドクと議論し研究内容について助言をもらうきっかけにな り、大きな研究機関に所属するメリットを感じることができた。また、日本から見ると「アメリカの研究者」と いう大きなくくりになりがちであるが、実際アメリカは広い。ニューヨーク州北部を中心とした、アメリカ北 東部の進化生物学者たちのローカルなつながりを感じるシンポジウムでもあった。 進化学研究は、近年の塩基配列決定技術の大規模化、それによるゲノム/配列情報リソースの飛躍的増 大によって、さらなる発展の機会を得たと期待されます。そこでこの連載では、進化研究に有用と思われる ツール/ソフトウェアを紹介していきたいと思います。主に扱う内容は DNA/RNA配列情報解析となります が、それ以外でも有用なものは採りあげたいと考えます(例えば種多様度解析のソフトウェアなど)。適宜リ クエスト下さい。また紹介にあたっては、出来るだけツール/ソフトウェア論文の著者ご自身に登場頂き、開 発の動機・背景や使用法の解説など、論文だけでは手に入らない生き生きとした情報をお伝えできればと思 います。
第一回目は、RNA sequencing(RNA-Seq)データから高精度で発現量を推定するソフトウェア TIGAR を 出版された東北大学・助教の成相直樹さんに、執筆頂きました。RNA-Seqは、進化学でしばしば対象とする 非モデル生物、すなわちリファレンスゲノム配列がまだ決定されていない生物種において、網羅的な機能遺 伝子配列やその発現量の推定を可能にする手法で、進化研究でも大いに活用されつつあります。国内でも、 エゾサンショウウオの表現型可塑性や、爬虫類(カメ類)の異種間比較などの興味深い課題に適用した研究者 がおられます。そのようなRNA-Seqデータ、とくに発現量の解析では、バイアスや誤推定を低減し、実際の 生命現象を反映した高精度な定量化を実現することが、意義のある知見を得るための肝となります。成相さ んによる以下のTIGARの記事では、RNA-Seqの概論、変分ベイズ推定の導入による高精度化へのアプロー チ、現在の定番ツールであるTopHatおよびCufflinksとの性能比較、実際の使用方法(コマンド)などを解 説します。現在RNA-Seqを活用している方だけでなく、広く今後の研究内容を検討されている方に、有益な ヒントとなりましたら幸いです。
新シリーズ「大量データと知見の架け橋」
緒言・編集担当 佐藤行人
(東北大学 東北メディカル・メガバンク機構)July 2014 10 第 1回﹁ RNA -Seq による網羅的トランスクリプトーム解析﹂ はじめに 近年、次世代シークエンサ(NGS)によるRNA-Seq技術により、細胞の遺伝子発現を網羅的かつ一塩基レ ベルの高解像度で解析することが可能となった。これまで一般的に行われてきているマイクロアレイ技術を 利用した遺伝子発現解析においては、Agilent社、Affymetrix社等が提供する解析ソフトウェアを使用した り、RのBioconductorパッケージ等を使用したりすることが比較的簡単に出来る様になっている。しかしな がら RNA-Seqデータ解析については、新しいシークエンス技術の登場と共に様々な解析手法が日々提案さ れている。また、ツールごとに必要なマシンの環境、使用方法、出力結果の解釈等が異なることもあり、特 にウェットの研究者にとってRNA-Seqデータ解析の敷居が高いと感じてしまうことがあるかもしれない。今 回の解説記事ではRNA-Seqデータ解析をなるべく身近に感じてもらえるように、RNA-Seqデータ解析を行 う前の事前準備から、リードのアライメント(マッピング)方法、アライメント結果からの遺伝子発現レベル の正規化、定量まで、最も基礎的かつ重要なステップを解説したい。また、具体的にいくつかの既存ツール のアルゴリズム、長所・短所を概観する(表
1
)。本稿の目的は、RNA-Seqデータ解析が実は非常にシンプル であり、かつ応用範囲が広いことを認識していただくことである。RNA
-Seq
の特徴・利点 RNA-Seqは細胞から抽出したmRNAをcDNAに逆転写してPCR増幅した後、数百bp程度に断片化し、 次世代シークエンサで各フラグメントの片側、もしくは両側から塩基を読む。通常、各フラグメントの両側 100-250 bp程度を読み取る(PacBio RSII などでの一分子長鎖配列解析については別の機会に触れる)。転 写量推定の最初のステップとして、読んだ配列(リード)をリファレンス配列(ゲノム配列もしくはcDNA配 列)にアライメントする。次に、そのアライメント結果に基づき、リードがどのリファレンス配列から出てい るかを判断し、それぞれの転写産物の遺伝子発現量を推定する。その際、真の遺伝子発現量はシークエンサ で読めたcDNA断片の数に比例する、という仮定に基づく。マイクロアレイによる発現量の定量方法が蛍光 色素の検出を基にしていることに対して、NGSによるRNA-Seqではリード数を基に遺伝子発現量を定量す ることから、リード数を十分読めば、マイクロアレイに比べてダイナミックレンジが広いことが報告されてい る(Wang et al., 2009)。また、cDNAプローブがデザイン済みであるマイクロアレイ解析とは異なり、転写産 物の配列を直接シークエンシングできること、そしてサンプルのゲノム多型(cDNA配列から間接的に、では あるが)、及びアリル特異的な遺伝子発現が検出できることもRNA-Seqの大きな利点である。本稿では特に、 遺伝子発現量の定量手法について解説する。 ツール 長 所 短 所 ウェブサイト Cufflinks 新規転写産物を推定できる モデル生物の解析のみ対応 http://cufflinks.cbcb.umd.edu/ RSEM 高速。非モデル生物の解析も可能 きない新規転写産物を推定で http://deweylab.biostat.wisc.edu/rsem/ TIGAR 高精度な遺伝子発現量推定。非モデル生物の 解析も可能 新規転写産物を推定で きない https://github.com/nariai/tigar/ 表1 各ツールの比較第
1
回
「
RNA
-
Seq
による
網羅的トランスクリプトーム解析」
成相直樹
(東北大学 東北メディカル・メガバンク機構 ゲノム解析部門)日本進化学会ニュース July 2014 11 第 1回﹁ RNA -Seq による網羅的トランスクリプトーム解析﹂
RNA
-Seq
における遺伝子発現量の正規化 RNA-Seqデータから転写産物ごとの遺伝子発現量を定量するためには、リファレンスcDNA配列にアライメントした総リード数、及び転写産物(cDNA)の長さで正規化した値、FPKM(Fragments Per Kilobase of
exon per million fragments mapped)が良く使われる(Trapnell et al., 2012)。例えば、ある転写産物にアラ
イメントされたリード数が3,000リード、cDNA全長が5,000 bp、リード総数が4,000万リードだとする。そう するとこの転写産物のFPKMは FPKM=3,000 /(5,000 / 1,000)/(40,000,000 / 1,000,000)=15.0 と計算される。以上の定義から明らかである様に、FPKMの正確な算出のためには、そもそもリードが正し く転写産物(cDNA)のリファレンス配列にアライメント出来ていることが前提である。しかしながら通常、 リードの長さは100-250 bpであり、シークエンス精度は向上しつつあるものの0.1% -1.0%程度の置換・挿入・ 欠失エラーを含むため、必ずしもリードが正しい転写産物のリファレンス配列にアライメントされるとは限 らない。更に、選択的スプライシングにより生成される同じ遺伝子由来の転写産物は当然ながら塩基配列が お互い非常に良く似ており、あるリードがどの転写産物から生成されたかを正しく判断することは必ずしも 容易ではない。また、ある転写産物についてパラログ、偽遺伝子の関係にある遺伝子から得られる産物とは リファレンス配列が似ているため、リードをリファレンス配列にアライメントする際には上記と同様、マルチ マップの問題が発生する。このような曖昧なアライメント候補が複数存在する場合、一つの解決方法として、 マルチマップリードを無視することにして、ユニークにアライメントされたリードのみを基にして遺伝子発現 レベルを定量する、という方法が考えられる。しかしながらこの方法では本来ならば考慮に入れるべきリー ドを捨ててしまっているため定量性が落ちることが知られており(Mortazavi et al., 2008)、更にはトリプレッ トリピート等のlow complexity sequenceを含むような転写産物の定量が難しくなると予想される。従って現 在、標準的に使用されているRNA-Seqデータ解析手法の多くはマルチマップリードを捨てるのではなく、有 効利用するアルゴリズムを実装している。以降、各ツールの特徴について概説する。
既存手法(
TopHat/Cufflinks
)最もポピュラーに使用されているRNA-Seqデータ解析手法の一つに、TopHat/Cufflinksが挙げられる (Trapnell et al., 2009, Trapnell et al., 2012)。TopHatはリードをリファレンスゲノムにアライメントするツー ルであり、Cufflinksはそのアライメント結果から各転写産物の発現量を推定するツールである。当然、リー ドはリファレンスゲノムのexon領域にしかアライメント出来ないため、アライメントの際にはexon/intron 領 域のアノテーション情報が必要となるが、その際に使用するgtfファイルなどもまとめてパッケージとして提 供されている(http://ccb.jhu.edu/software/tophat/)。 第一のステップとして、TopHatではリードをそれぞれ25 bp程度に断片化した後、マルチマップを許した 上でリファレンスゲノムにアライメントする(その際、bowtieというアライメントツールを使用する)。これに よりリード断片がリファレンスゲノムにおけるexon領域にマッピングされたり、されなかったりする。マッピ ングされなかったリード断片の前後にexon領域にマップしたリード断片があれば、そのunmappedリードは exonとexonのジャンクションに由来するリードであると推定される。このアルゴリズムにより、新規の転写 産物を推定することも可能となっている。 次のステップとして、CufflinksはTopHatのアライメント結果を入力として、転写産物ごとのFPKMを計 算する。リードが複数の転写産物にマルチマップしている場合、デフォルトオプションではマルチマップして いる転写産物に等分にリードを分配する(たとえば、1本のリードが2つの転写産物にアライメントしている 場合、それぞれの転写産物から0.5本ずつ出力された、と考える)。最新のバージョンでは -u オプションを 付けることにより、周辺のユニークマップしたリードの数に基づいて、マルチマップのリードを比例配分する
July 2014 12 第 1回﹁ RNA -Seq による網羅的トランスクリプトーム解析﹂ を25 bpの長さに断片化してリファレンスゲノムにアライメントするアルゴリズムを採用しているため、アライ メント自体の偽陽性を含むことは避けられず、結果として定量性が損なわれる、あるいは間違って新規の選 択的スプライシングを推定してしまう、といった可能性を常に留意しておく必要がある(この点については、 後述する)。 TopHat/Cufflinksの長所として、Cuffdiffなどいくつかの下流解析を行うツールがパッケージとして提供 されているため、とりあえず一連のRNA-Seq解析を行うことが出来ることが挙げられる。また、多くの研究 者が既に使用しているため、問題が発生したときに助けてくれる仲間が多いことが期待できる。逆に欠点と して、TopHat/Cufflinksはモデル生物(ヒト、マウス、ラット等)の解析をメインターゲットにしているため、 非モデル生物のトランスクリプトーム解析には向いていない。また、デフォルトオプションでは新規の転写産 物を予測してくれるものの、この予測精度については現時点では十分に検証されているとは言い難い(短い リード断片から新規の選択的スプライシングを予測することが難しいことは容易に想像できる)。新規の転写 産物を予測したくない場合は、TopHat/Cufflinksのオプションで -G オプションを付けることにより、既知 の転写産物のみの発現レベル推定を行うことも出来る。 統計的モデリングによるマルチマップリードの解決と転写量推定 RNA-Seqデータ解析において、リファレンス配列(ゲノム配列、もしくはcDNA 配列)にアライメントされ たリード数に基づいて転写量推定を行うこと、従ってリードが複数箇所のリファレンス配列にアライメントす る場合(マルチマップ)の扱い方が問題になることを説明した。前述の通り、TopHat/Cufflinksにおいては -u オプションを付けることにより、rescue methodと呼ばれるアルゴリズム(周辺領域のユニークマップリー ドの情報に基づく重み付け)によって、ある程度マルチマップの問題を解決している。しかしながらこのアル ゴリズムでは、マルチマップの問題を完全に解決しているとは言い難い。 近年、マルチマップリードのアライメントを隠れ変数として扱い、転写量をパラメータとして推定する統 計的な手法が提案されている(Li et al., 2010, Nariai et al., 2013)。説明のため、シンプルな例を挙げて説明 する。今、isoform A, B, Cという転写産物が発現しているとする。それぞれのリードがどの転写産物から生 成されたものであるか、リファレンスcDNA配列にリードをアライメントすることにより同定する。まず簡単 のため、アライメントは一意に決まるとする。それぞれの転写産物について、アライメントされたリード数に 基づいて、転写量(ここではリードの割合)を推定することができる(図
1
)。アライメントが一意に決まらな い場合(マルチマップ)、統計的枠組みの下、アライメントを隠れ変数として推定することが出来る(ここで は詳細な説明は省く)。この様な統計的手法の利点として、シークエンスデータ(リード)自体のエラーをモ デリングできること、そしてデータの生成モデルを仮定した上で統計的に尤もらしいアライメントと転写量を 同時に推定できることが挙げられる。このような統計 的手法を適用することで、シークエンシングエラーや アライメントエラーなどに強い、ロバストな転写量推 定が可能となる。パラメータ推定はEMアルゴリズム (もしくは変分EMアルゴリズム)により、隠れ変数の 推定とパラメータのアップデートを交互に繰り返すこ とで、解析的に(局所)最適解が得られる。以下、著 者らが開発した統計的手法であるTIGAR(Transcriptisoform abundance estimation method with gapped alignment of RNA-Seq data by variational Bayesian inference)の利点について解説する。 RSEM(Li et al., 2010)においては、リードデータ は挿入・欠失を含まないことを前提としてモデリング 図1 リードをリファレンス cDNA 配列にアライメ ントすることにより、それぞれの転写量を推定する ことが出来る
日本進化学会ニュース July 2014 13 第 1回﹁ RNA -Seq による網羅的トランスクリプトーム解析﹂ されている。この仮定は、HiSeqやMiSeqなど、Illumina社のシークエンサ機器から得られる100 bp程度の ショートリードに関しては挿入・欠失エラーが比較的少ないため、大きな問題とはならないかもしれない。し かしながらLife Technologies社のIonPGM、Pacific Biosciences社のRS IIなどの比較的、挿入・欠失エ ラーを多く含むシークエンスデータに対しては、必ずしも適切では無い。筆者らが開発したTIGARにおいて は、挿入・欠失エラー、すなわちリファレンスcDNA配列に対するギャップ付きアライメントの状態をPair HMMという確率モデルを利用することで表現し、データの尤度を計算する。この様にギャップ付きアライメ ントを受け付けるモデルを採用することにより、bowtie2、bwa-mem、novoalignなどの高感度なアライメン トツールが利用できる(これに対して、RSEMはギャップ付きアライメントを行わないbowtieを使用する)。 さて、上述の通り、TIGARではリードのアライメントの際、ギャップ付きアライメントを行うことで感度 を向上することが出来る。しかしながら、同時にアライメントの偽陽性についても注意を払わなければならな いことは前項のTopHatのアルゴリズム解説の中でも述べた。RSEMにおいては、EMアルゴリズムによっ て隠れ変数(アライメント)の推定とパラメータ(各転写産物の転写量)の最尤推定を行う。これに対して、 TIGARは、変分ベイズ推定によってパラメータを事後分布として推定する。ベイズ推定ではパラメータを点 推定ではなく事後分布として推定するため、ノイズに強いロバストな推定を行うことが出来る。また、事前分 布のハイパーパラメータの設定により、モデルの複雑さ(この場合は推定されるべき転写産物の数)をコント ロールできるという特徴がある。 ここで、なぜ 推定されるべき転写産物の数 を気にする必要があるかについて述べる。極端な例を挙げる と、ある遺伝子座位について、exonの数をnとすれば、選択的スプライシングにより生成可能な転写産物の 組み合わせの数は理論的には2n種類が可能であり、これを全ての遺伝子座位について考えると途方も無い転 写産物候補を仮想的には考えることが可能である。また、融合遺伝子の可能性についても考えると、更に膨 大な候補数となる。しかしながら、通常、ある細胞のある時点においては、これら膨大な候補のうち、せい ぜい数千∼数万程度が実際には遺伝子発現していると考えるのが自然である。TIGARは、ベイズ推定の枠組 みの下、より少ないパラメータ数(転写産物の候補の数)でデータを説明するモデルを選択するという オッ カムの剃刀 の精神に従うようなパラメータ推定を行うことが出来る(図
2
)。より詳細なアルゴリズムについ ては(Nariai et al., 2013)を参照していただきたい。TIGAR
の使用方法 TIGARを使用する一連の解析の流れをまとめてパイプラインと呼ぶことにする。パイプラインの構成要素 図2 オッカムの剃刀の概念図。データをより少な い仮定で説明できれば、それに越したことは無い、 とする精神(http://ja.wikipedia.org/wiki/オッカム の剃刀) 図3 TIGAR のパイプラインJuly 2014 14 第 1回﹁ RNA -Seq による網羅的トランスクリプトーム解析﹂ は、1)RNA-SeqリードのリファレンスcDNA 配列へのアライメント、2)アライメント結果からの転写量推定、 の2つに大きく分けられる(図
3
参照)。 ここでリファレンス配列はcDNA配列であり、例えばマウスのcDNA配列であれば以下の様にUCSCの ウェブサイトからダウンロードできる: wget http://hgdownload.soe.ucsc.edu/goldenPath/mm9/bigZips/refMrna.fa.gz リードデータ(FASTQ)を上記で得られたリファレンスcDNA配列にアライメントする際には、ギャップ付 きアライメントを行うことができる bowtie2、あるいはbwa-memなどを推奨している。いずれのツールを使 う場合でも、最初に一度だけ、リードの高速なアライメントのためリファレンス配列のFMインデックスを作 成する必要がある(FMインデックスの詳細はLi and Durbin, 2009を参照):mkdir ref
bowtie2-build index refMrna.fa ./ref/refMrna
FMインデックスを作成後、マルチマップを許したアライメントを行う: シングルエンドデータの場合:
bowtie2 -k 100 --very-sensitive ./ref/refMrna sample.fastq > sample.sam ペアエンドデータの場合:
bowtie2 -k 100 --very-sensitive ./ref/refMrna -1 sample_1.fastq -2 sample_2.fastq > sample.sam
最後に、TIGAR本体のプログラムを実行する:
シングルエンドデータの場合:
java -jar Tigar.jar refMrna.fa sample.sam --alpha_zero 0.1 sample_out.txt ペアエンドデータの場合:
java -jar Tigar.jar refMrna.fa sample.sam --is_paired 1 --alpha_zero 0.1 sample_out.txt
上記の様に、cDNAリファレンス配列さえ準備できれば、TIGARは非常にシンプルなパイプラインで実行 することが出来る。これはすなわち、非モデル生物のRNA-Seqデータ解析も同様のパイプラインで実行可 能であることを意味する。尚、プログラム、実行マニュアルはプロジェクトのウェブサイトから入手できる (https://github.com/nariai/tigar)。
TIGAR
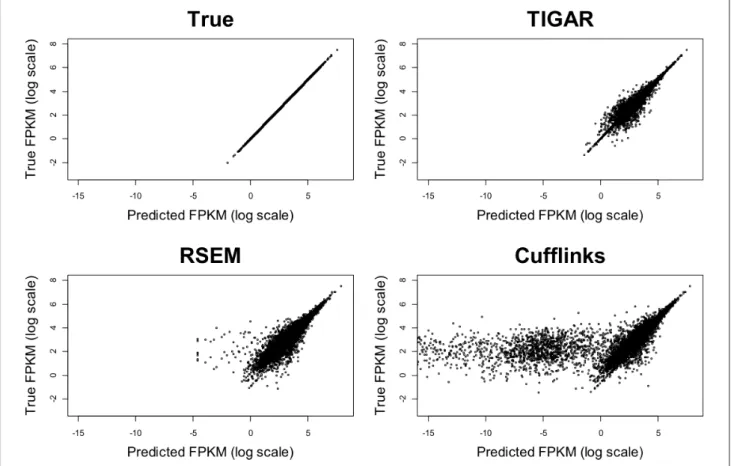
と既存ツールのパフォーマンス比較 ここではシミュレーションデータを使用することで、TIGARと既存ツール(TopHat/Cufflinks、RSEM) とのパフォーマンス比較を行う。RefSeqのデータベースからヒトのcDNA配列をランダムに10,000個選び、 それぞれの遺伝子発現量が対数正規分布に従っていると仮定して 正解の FPKMを設定する。ここでは入 力データとして、100 bp x 2(ペアエンド)の人工的なRNA-Seqデータを100万リード生成した。シークエンサ のエラーとして、置換・欠失・挿入エラーを1%導入する。それぞれのソフトウェアで転写量推定を行って、 真の転写量との相関を見た(図4
)。転写量が大きい転写産物(たとえばFPKM>50)はどのツールでも上手 く推定できるが、転写量が小さい転写産物は転写量の定量がRSEM、TopHat/Cufflinksでは難しくなること が分かる。この理由として、既存ツールがリードのアライメントを間違えている、あるいはマルチマップリー ドを適正に配分できていないため、結果として正確な転写量を推定することが出来ていないことが考えられ日本進化学会ニュース July 2014 15 第 1回﹁ RNA -Seq による網羅的トランスクリプトーム解析﹂ る。一方、TIGARでは実際の転写量と良く相関した転写量推定が出来ていることが分かる。今後、single-cell sequencingデータへの適用等が考えられる。 まとめ 本稿ではRNA-Seqデータから転写量推定を行う一般的な流れと、既存ツールのアルゴリズム紹介、筆者 らが開発した統計的手法TIGARの具体的な使用方法、及びシミュレーションデータによる実行結果の例を紹 介した。RNA-Seqデータを解析する際の基本的な考え方、注意点などを理解していただくことが出来たのな ら、幸いである。スペースの関係上、新規の選択的スプライシングの検出方法については詳細な解説を行わ なかったが、興味のある読者は参考文献(Mezlini et al., 2013)等を参照していただきたい。また、通常、転 写量推定を行った後、発現差解析、パスウェイ解析などの下流解析に進むのが一般的である。これらのト ピックについても今回は省略する。興味ある読者にとって東京大学・門田幸二先生のウェブサイト(http:// www.iu.a.u-tokyo.ac.jp/~kadota/r_seq.html)が参考になると思われる。 参考文献
[1] Wang et al., Nat Rev Genet. (2009) 10:57-63 [2] Mortazavi et al., Nat Methods. (2008) 5(7):621-628. [3] Trapnell et al., Bioinformatics. (2009) 25(9):1105-1111. [4] Trapnell et al., Nat Protoc. (2012) 7(3):562-578. [5] Li et al., Bioinformatics. (2010) 26(4):493-500. [6] Nariai et al., Bioinformatics. (2013) 29(18):2292-2299. [7] Li and Durbin, Bioinformatics. (2009) 25(14): 1754-60. [8] Mezlini et al., Genome Res. (2013) 23(3):519-29
July 2014 16 第 18回海外研究室だより ジュネーブからケンブリッジへエピジェネティクス研究室紹介 16 私は現在イギリスのケンブリッジ大学、 Sainsbury Laboratoryでポスドクをして います。私の研究テーマは植物のエピジェ ネティクスで、もう少し詳しく言うと、世 代を超えて遺伝するエピジェネティックな 分子機構やトランスポゾンの制御機構につ いて研究しています。エピジェネティクス という分野にそれほど馴染みのない方もい らっしゃるかと思いますので、少しだけこ こに紹介します。 エピジェネティクスとは、DNA配列の変 化を伴わない遺伝子発現変化を研究する学 術分野の事であり、その基盤となる分子機構としては、DNAメチル化やヒストン修飾といった化学修飾が深 く関わっています。エピジェネティックな遺伝子発現の変化は細胞分裂を経ても維持され、さらに世代を超 えて遺伝するものもあります。エピジェネティックな制御は、発生分化や環境応答、トランスポゾンのサイレ ンシング等、様々な局面において関わっていますが、近年、環境要因によって生じたエピジェネティックな変 化が次世代に遺伝したという、獲得形質の遺伝を示唆する報告も多くの生物種において報告されており、進 化学的にも注目を集めています。しかし、この獲得形質の遺伝については、実際に多くの報告がなされてい ますが、特に植物においてはその分子機構は解明されておらず、その再現性や安定性についても議論の分か れるところです。適応戦略としての世代を超えたエピジェネティックな形質の遺伝については、もたらされる 利益の他に、被るかもしれない不利益についても考慮する必要があります。例を挙げて言うと、あるストレス に対して変化した形質が世代を超えて記憶され続けるという事は、後世代において状況に応じた適切なスト レス反応を阻害するかもしれないということです。私のこちらでのプロジェクトとして、 ストレスにより生じ たエピジェネティックな変化の遺伝を妨げるシステムが存在するかについて、遺伝学的スクリーニングを用い て研究し、実際に植物にはそのようなシステムがある事を発見しました。このシステムには二つの遺伝子が関 わっているのですが、そのうちの一つの遺伝子は動物においても高度に保存されており、動物においても同 様なシステムが働いているかどうかも興味深い所です[1]。 世代を超えて遺伝するエピジェネティック修飾の一つにDNAメチル化があります。DNAメチル化は哺乳 類と植物で観察されますが、哺乳類では胚発生時にDNAメチル化の大部分がリセットされるのに対し、植物 においては何世代にもわたり非常に安定に維持されるため、エピゲノムのバリエーションを進化学的な観点 から研究する際にも、有用な材料であると言えます。エピゲノムの変化によって生じる表現型は、自然界に おいても花や果実の色・模様、遺伝的不和合性など多々見られます。 これらのエピゲノム変異の起源につい ては定かではなく、環境要因がエピゲノムのバリエーションに寄与するかどうかについてはまだ議論の余地が 多い所です。この問題に関与する一つの因子として、トランスポゾンの転移があります。トランスポゾンは菌 類、動物、植物とあらゆる生物種に広く存在し、ゲノムの主要な構成要素になっています。大部分のトラン スポゾンは通常エピジェネティックな制御によりサイレンシングされていますが、様々なストレス刺激はこれ らのトランスポゾンを活性化させ、時には転移を引き起こすことが知られています。転移したトランスポゾン
岩崎まゆみ
(ケンブリッジ大学、Sainsbury Laboratory)第
18
回
海外研究室だより
ジュネーブからケンブリッジへ:
エピジェネティクス研究室紹介
図1 植物園に隣接するケンブリッジのSainsbury Laboratory。 モダンで美しい建物は数々の建築賞を受賞しています。日本進化学会ニュース July 2014 17 第 18回海外研究室だより ジュネーブからケンブリッジへエピジェネティクス研究室紹介 17 はしばしば近傍の遺伝子もエピジェネティックな変化を引き起こします。B. McClintockは トランスポゾン の活性化はゲノムを再編する事で環境に適応する機会を与える、適応反応の一つである と述べましたが[2]、 彼女の仮説を裏付けるように、多くの研究がトランスポゾンがゲノムの進化や環境適応に一定の役割を果た している事を示しています。トランスポゾンの制御機構や宿主ゲノムとどのように相互作用し進化して来たか についてはまだ不明な点も多く、今後の研究展開が期待されるところです[3]。 留学生活について
私は以前スイスのジュネーブ大学に所属していましたが、2013年の秋に私のボスであるProf. Jerzy
Pasz-kowski(a.k.a Jurek)の異動に伴ってケンブリッジに引っ越して来ました。そこでスイスとイギリス、二つの 国での生活環境とラボの引っ越し体験についても紹介しようと思います。 まずスイスの都市部で生活を始めるにあたり、ほとんどの人にとって問題になるのが住宅問題だろうと思 います。特にジュネーブの賃貸住宅事情は最悪で、家賃は極めて高い上に空き家率はスイスの中で最低で (0.2%程度)、一旦空き部屋情報が出ると申込者が殺到します。申込者の選定は、オーナーもしくはRegieと 呼ばれるアパート管理会社によって行われますが、フランス語を話さない外国人の優先順位は通常低く置か れるため、私たち日本人にとってアパート探しは更に困難なものになります。私は幸運にも、ちょうど入れ違 いで日本に帰国する事が決まっていた同じラボのポスドクの方の部屋を引き継ぐことが出来ましたが、半年、 時には一年以上アパート探しに明け暮れるという話は珍しくもありません。結局住む所が見つからず、留学を 断念して帰国した人の話も聞きました。 しかし住宅問題を除けば、スイスの生活水準は総じて高く、治安も良いため、とても暮らしやすい国だと思 います。また大自然に囲まれており、ウィンタースポーツはもちろん、ハイキング・トレッキングコースも非 常に良く整備されており、これらのアクティビティが好きな人にとってはとても魅力的な所です。スイスは大 まかに、ドイツ語圏、フランス語圏、イタリア語圏と3つの言語圏に分けられます(ロマンシュ語という4つ目 の公用語がありますが、母語人口は非常に少なく絶滅が危惧されています)。各言語圏はそれぞれ隣り合った 国、つまりドイツ、フランス、イタリアから文化的な影響を受けており、その文化の違いはかなり大きいよう にみえます。 ちなみにそれぞれの文化圏の障壁を表す用語として、「レシュティ(スイスのジャガイモ料理)の 溝」(ドイツ語:Röstigraben、フランス語:barrière de Rösti)という言葉が存在するようです。フランス語圏 であるジュネーブは、個人主義、自由主義的な思想が浸透しているように感じますが、保守的で何事にもきっ ちりしているドイツ語圏と比べてみると、人種は多様で街はゴミゴミしており、役所仕事や事務仕事等も結構 適当にこなされている感が強いです。しかし、この自由な雰囲気は私にとっては居心地のよいものでした。 さてスイスの研究環境はというと、 研究資金は潤沢で、基礎研究を重視しており、研究水準も非常に高く、 とても恵まれた環境だと言えるでしょう。後の要素はラボによって異なってくると思いますが、私の所属して いるラボの特徴は、一言で表すと、とても自由度の高いラボで、これもまた私に取っては大変ありがたいもの 図2 ジュネーブ近郊、レマン湖畔にあるJurek(写真左)のお宅にて。ひと泳ぎした後バーベキューを楽しみました。