15
hp160087 「京」産業利用(実証利用) K Industrial Use
Computational Study of Electrical Conductivity Properties of Organic
Semiconducting Polymers and Metal Complex-host Systems
高分子有機半導体材料及び金属ホスト系
の電気伝導特性の計算科学的研究
Masaya Ishida1), Michiaki Arita1), Shinya Nishino1) and Takeo Hoshi2)
石田 雅也1)、有田 通朗1)、西野 信也1)、星 健夫2)
1) Sumitomo Chemical Co., Ltd., 2) Tottori University 1) 住友化学株式会社、2) 鳥取大学
Abstract
We analyzed the carrier transfer (CT) among oriented polymer chains of organic semiconductor materials by the wave packet dynamics (WPD) implemented in ELSES, an extra large-scale electronic structure calculation code. The characteristic vibration in CT can be understood by the resonance states in a two-level system. The WPD was applied to metal complex-host systems. The hole mobility was calculated for single-crystalline anthracene, a host system of an organic molecular assembly. We verified the accuracy of the tight-binding (TB) parameters of organometallic complexes and performed benchmarks for large-scale calculations connected to a basic study of WPD in metal complex-host systems.
Keywords: Organic Semiconducting Polymer, Fluorene, ELSES, Anthracene, Ir(ppy)3, CBP, Electron
Conductivity, Wave Packet Dynamics 要旨 超大規模電子状態計算手法 ELSES に実装された波束ダイナミクスの計算を用いて、高分子有 機半導体材料における配向構造を有する高分子鎖間の電荷伝播挙動を解析した。電荷伝播で見ら れた特徴的な振動構造は、2 準位系の共鳴状態で説明できることが分かった。この波束ダイナミ クス計算を金属錯体-ホスト系のシミュレーションへ発展させるべく、有機低分子集合体であるホ スト系のモデルとして採用したアントラセン単結晶モデルに対して波束ダイナミクス計算を適 用しホール移動度を算出した.さらに,有機金属錯体のTight Binding(TB)パラメータの検証と大 規模計算のベンチマークを通して、金属錯体-ホスト系における波束ダイナミクス計算の基礎検討 を実施した。
© 2021 Research Organization for Information Science and Technology All rights reserved. Received: 20 April 2020
Accepted: 2 April 2021 Available online: 28 April 2021
16 Fig. 1 Side view (left) and top view (right) of
(4×4×4) superlattice of crystalline anthracene.
Fig. 2 Structure of Ir(ppy)3+ (CBP)99.
キーワード:高分子有機半導体、フルオレン、ELSES、アントラセン、Ir(ppy)3、CBP、電気伝導、
波束ダイナミクス
1. Background and Objectives of Research
Organic semiconductor materials are crucial for next-generation Internet-of-Things (IoT) products, such as flexible devices. It is therefore of great importance to understand their functions and the carrier transfer (CT) mechanism for materials design. The present paper investigates the conduction mechanism from the viewpoint of the electronic states in aggregate structures of organic polymers using the ELSES code [1] developed by Hoshi et al., to inform materials design. We study a single crystal of anthracene and a metal complex-host, to confirm that the method is applicable to aggregated small molecules as well as polymers.
2. Calculation Model
A. Fluorene aggregate model
An aggregate structure model was built from three oriented fluorene (FL) decamers with octyl groups at the 9-position. The model is called model 𝛽𝛽 in Fig. 4 (a) of Ref. [2] and will be presented later in this paper. A wave packet dynamics (WPD) simulation was carried out within a quantum molecular dynamics (QMD) simulation using the tight-binding approximation [2], where the eigenstates are calculated at each time step and the wave packet is described as a linear combination of the eigenstates.
B. Anthracene single-crystalline structure model
An anthracene single-crystalline structure model was built as part of a preliminary study of the complex-host material in Sec. 2-C. A 4×4×4 supercell, shown in Fig. 1, was used for the periodic simulation cell.
C. Iridium complex-host material model
Several molecular assemblies were built from the metal complex Ir(ppy)3 and the host molecule 4,4′-bis
(9H-carbazol-9-yl) biphenyl (CBP). The system contains approximately N=32,000 atoms. Fig. 2 shows the structure of Ir(ppy)3 + (CBP)99, one of our molecular assemblies.
17
Fig. 4 WPD for oriented aggregate structures. (a), (b):
Time-evolution of the Mulliken charges 𝜌𝜌3, 𝜌𝜌2 on the polymer
chains β3, β2, respectively, (c), (d): Wave packet at t = 1.0 ps
and 1.9 ps, respectively.
Fig. 3 Benchmark with the (CBP)100 system in (a) our in-house
computer and (b) the K computer.
3. Method and Performance of Parallel Computing
Benchmark tests were carried out with up to 48 MPI processes on our in-house simulator and 2,048 MPI processes (2,048 nodes) on the K computer. Fig. 3 plots the elapsed time per molecular dynamics (MD) step 𝑇𝑇𝑝𝑝 with 𝑃𝑃 processors for the (CBP)100 system.
Reasonably strong scaling is seen
in both cases. The parallel efficiency 𝛼𝛼𝑃𝑃 is defined as 𝛼𝛼𝑃𝑃 ≡ 𝑇𝑇1 / (𝑃𝑃 𝑇𝑇𝑃𝑃) for the in-house computer, and we obtained 𝛼𝛼48= 0.71. The parallel efficiency on the K computer 𝛼𝛼𝑃𝑃 is defined as 𝛼𝛼𝑃𝑃 ≡ 128𝑇𝑇128 / ( 𝑃𝑃 𝑇𝑇𝑃𝑃) and we obtained 𝛼𝛼256= 0.89, 𝛼𝛼512 = 0.79, 𝛼𝛼1024= 0.53, 𝛼𝛼2048= 0.35.
4. Research Results
A. Fluorene aggregate model
In general, the CT in organic molecular assemblies can be split into intra-chain and inter-chain conduction, and the mechanism is closely related to the aggregated structural features [3]. In particular, inter-chain conduction or hopping conduction plays an essential role in the CT of organic semiconductors [4].
We analyzed the CT behavior of the two FL models, models 𝛼𝛼 and 𝛽𝛽, in different aggregated structures by WPD calculations [2]. The WPD result for model 𝛽𝛽 is shown in Fig. 4. The three polymer chains in the model are denoted as β1, β2, and β3.
The initial wave packet is chosen as
18
Fig. 5 Participation ratio (PR) of the system.
chain β1 to β3. In the current study, we examine the transfer from β1 to β2, which was not examined in the
previous study. The Mulliken charges on β1, β2, and β3 are denoted as 𝜌𝜌1, 𝜌𝜌2, and 𝜌𝜌3, respectively, and
satisfy the relation 𝜌𝜌1+ 𝜌𝜌2+ 𝜌𝜌3= 1.
Figs. 4 (a) and (b) show the time-evolution of 𝜌𝜌3 and 𝜌𝜌2, respectively. Figs. 4 (c) and (d) visualize the WPD at t = 1.0 and 1.9 ps. Figs. 4 (a) and (b) show the inter-chain CT from β1 to β3 and β2. The vertical
axes of Figs. 4 (a) and (b) are log-scaled, and thus show that the hopping from β1 to β2 is negligibly small
compared to that from β1 to β3. This is because the chains of β1 and β2 are well separated and the overlap of
atomic orbitals between them is small. Hence the CT from β1 to β2 is invisible in Figs. 4 (c) and (d) and the
CT to β2 is invisible in Fig. 5 (g) of Ref. [2].
The characteristic vibrational behavior of the inter-chain hopping from β1 to β3 can be understood as
that of two-level resonant states [2]. The two levels here correspond to the HOMO levels h11 and h33 of the
polymer chains β1 and β3, respectively. Suppose that the inter-chain hopping is an off-diagonal element h13
of the Hamiltonian. Then, we can approximately describe the oscillator period as 𝜏𝜏 = 2𝜋𝜋/𝐸𝐸 (a.u.), where τ = 1 time a.u. is about 1/40 fs. If 𝐸𝐸 ≈ 0.001 a.u., the oscillation period is 2𝜋𝜋/𝐸𝐸 (a.u.) = 2π/0.001 a.u. ~ 6000/40 fs ~ 102 fs, which is comparable to the vibration period seen in Fig. 4 (a). Therefore, the energy
scale is around | h11-h33 | ~ | h13 | ~ E ~ 0.001 a.u.. From ELSES, the HOMO levels of β1 and β3 are 11.63 and
11.68 eV, respectively. Thus, the energy difference is around 0.055 eV or 0.002 a.u.. The above analysis quantitatively explains the CT behavior as being due to inter-chain hopping of pico-second order.
B. Anthracene single-crystal structure model We equilibrated the system from an initial structure
in Fig. 1. First, a rough equilibration was performed with the OPLS-AA force field by LAMMPS [5]. Then, the system was further equilibrated by the QMD by ELSES [1]. Each equilibration process was performed in a canonical ensemble at 300 / K. After the equilibration, we carried out a WPD calculation and calculated the carrier mobility. We also calculated the participation ratio (PR) [6] of the system. Fig. 5 depicts the PR of each energy level. The PR represents
the spatial locality of the eigenstates. Since holes work as a carrier in anthracene, the wave packet can be considered as a linear combination of the localized eigenstates below the HOMO level.
The WPD in ELSES can take into account the time-evolution of wave packets during the thermal atomic motion. Atomic oscillation occurs due to thermal fluctuations, and triggers carrier conduction by working as a perturbation to the wave packet. To evaluate the carrier mobility, we need to calculate the
19
Fig. 6 MSD of the wave packet as a function of
the simulation time. mean square displacement (MSD) of the wave packet
from its center of mass. The MSD shows a linear behavior with respect to the simulation time, and its derivative corresponds to the diffusion coefficient. A prefactor to the diffusion coefficient gives the carrier mobility by Einstein’s relation. Fig. 6 shows the time-evolution of the MSD in this study. From this, the carrier mobility was estimated to be 0.4 / cm2V-1s-1.
The experimental values [7] are in the range of 0.57– 2.07 / cm2V-1s-1, and thus we can conclude that the
calculated value is quantitatively reliable.
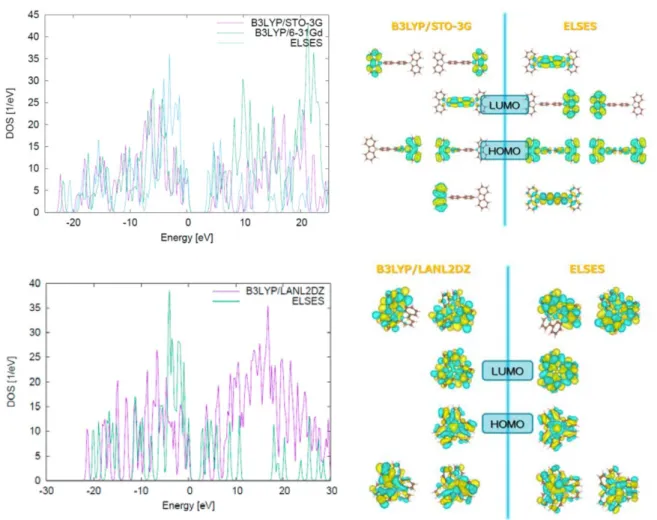
Fig. 7 DOS and MOs of CBP (upper) and Ir(ppy)3 (lower).
C. Iridium complex-host material model
As basic research, we verified the accuracy of the TB parameters with Ir(ppy)3 and CBP monomers. We
compared the density of states (DOS) and molecular orbitals (MOs) of each monomer with first-principles calculations by Gaussian (B3LYP/STO-3G, B3LYP/6-31G (d, p), B3LYP/LANL2DZ) and ELSES. For Ir, the TB parameters were taken from Ref. [8]. The other elemental parameters used default values [1]. Fig. 7
20
shows the DOS and MOs of Ir(ppy)3 and CBP monomers, respectively. The DOS is well reproduced for
both CBP and Ir(ppy)3, but ELSES underestimates the HOMO–LUMO gap by around 1 eV. For CBP, the
degenerated HOMO states are well reproduced, while the LUMO is exchanged with the adjacent level. We can say, however, that the energy difference in these levels is small and will not cause temperature fluctuation problems in the MD. On the other hand, the HOMO and LUMO for Ir(ppy)3 remain consistent.
Thus, ELSES is expected to be sufficiently accurate for MD and WPD.
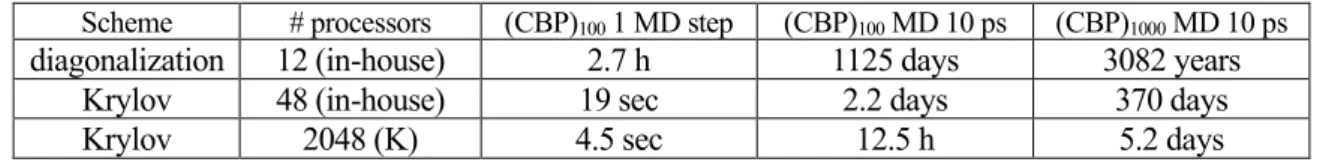
Table I shows benchmark results with our in-house computer and the K computer. We compared the diagonalization and the Krylov subspace method. The results show that the K computer is indispensable for multiple simulations of hundred-molecule systems with different conditions of temperature and concentration of the complex, and for analyzing larger systems in a realistic time.
Table I Benchmark results for an in-house computer and the K computer
Scheme # processors (CBP)100 1 MD step (CBP)100 MD 10 ps (CBP)1000 MD 10 ps diagonalization 12 (in-house) 2.7 h 1125 days 3082 years
Krylov 48 (in-house) 19 sec 2.2 days 370 days
Krylov 2048 (K) 4.5 sec 12.5 h 5.2 days
5. Summary and Future Subjects
Using WPD implemented in ELSES, we analyzed the CT behavior of oriented polymer chains. The characteristic CT behavior can be explained by the resonance states of a two-level system. The hole mobility of crystalline anthracene was calculated to be comparable to the experimental results, indicating that the evaluation of mobility by WPD is useful for crystalline organic semiconductors. Furthermore, we verified the accuracy of the TB parameters and performed benchmarks for large-scale calculations for metal complex-host materials, which opens the door for practical and accurate simulations on complex-host materials in the near future.
References
[1] http://www.elses.jp; T. Hoshi, et. al, J. Phys.: Condens. Matter 24, 165502/1-5 (2012) [2] M.Ishida, S.Nishino, Y. Abe and T. Hoshi, HPCI Research Report 4, 37 (2019). [3] M.Ishida, Y.Kurita and A.Nakazono Sumitomo Kagaku 2015, 25.
[4] N.Okabayashi, et al., Phys. Rev. Lett. 104, 077801 (2010). [5] http://lamps.sandia.gov
[6] T. Fujiwara, T. Mitsui and S. Yamamoto, Phys. Rev. B 53, R2910 (1996).
[7] R.G.Kepler, Phys. Rev. 119, 1226 (1960); http://www.rsi.co.jp/kagaku/cs/news/pdf/adf/201107.pdf [8] Y. Tsuji, R. Hoffmann and J. S. Miller, Polyhedron 103, pp. 141–149 (2016).