糖化産物
dihydropyrazinesによる炎症作用の抑制
江﨑 円香
Dihydropyrazine suppresses inflammation
Madoka Esaki March, 2021
Dihydropyrazine suppresses inflammation
Madoka Esaki
Dihydropyrazines (DHPs) are glycation products generated by the Maillard reaction, in which the carbonyl group of a reduced sugar binds to an amino residue of a protein in a nonenzymatic fashion. Glycation products are constantly generated and accumulated in our bodies and they can trigger chronic diseases, such as complications of diabetes, atherosclerosis, and neurological disorders. In contrast, some glycation products show antioxidant and antimutagenic effects that are beneficial to health. Therefore, it is important to determine and categorize the biological effects glycation products. We have focused on 3-hydro-2,2,5,6- tetramethylpyrazine (DHP-3). DHPs are generated from a dimer of D-glucosamine and 5-aminolevulinic acid. Methyl-substituted DHPs can generate several kinds of radicals that can result in DNA strand-cleavage. We previously revealed that DHP-3 activates the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway in HepG2 cells. Nrf2 responds not only to oxidative stress but also to inflammation. Thus, this study investigated whether DHP-3 affects Toll-like receptor 4 (TLR4)-driven inflammation.
1) DHP-3 suppresses activation of the TLR4 pathway in LPS-stimulated cells.
TLR4 recognition of lipopolysaccharide (LPS), a membrane glycolipid of Gram-negative bacteria, evokes inflammation. Inflammation via the TLR4 pathway is involved in various diseases, such as sepsis, hepatitis, and carcinoma. In this study, we examined the effects of DHP-3 on TLR4 signaling in cultured cells (HepG2, J774.1, and HL60 cells). The cells were stimulated with LPS and further exposed to DHP-3. Expression levels of factors in the TLR4 pathway were analyzed by Western blotting and/or q-PCR. First, we examined TLR4, which is at the start of the inflammatory signaling pathway. DHP-3 suppressed expression of TLR4 at both mRNA and protein levels. DHP-3 also reduced the levels of the TLR adaptor protein, MyD88.
Additionally, DHP-3 decreased the level of phosphorylation of each downstream factor in the TLR4 pathway, IκB, NF-κB, ERK, JNK, and p38. DHP-3 also suppressed NF-B nuclear translocation and c-Jun protein levels. These results indicated that the anti-inflammation effect mediated by DHP-3 resulted from suppression of the TLR4 pathway.
2) DHP-3 suppresses production of inflammatory mediators.
Cytokines, such as interleukins, chemokines, and interferons, are inflammatory mediators that play important roles in maintaining homeostasis, and numerous diseases result from disruption to cytokines. In this study, we show that DHP-3 suppressed inflammatory mediators in LPS-stimulated cells. LPS stimulation induced the chemokine CCL2, and pro-inflammatory cytokines, TNF, IL-1 and IL-6. This upregulation was abrogated by DHP-3 exposure. We also investigated the effects of DHP-3 on eicosanoid production. Prostaglandin and thromboxane are major eicosanoids generated from membrane lipid by cyclooxygenase (COX) that regulate inflammation. DHP-3 tended to attenuate expression levels of COX2, which suggested that DHP-3 suppressed eicosanoid production. These results indicate that DHP-3 suppressed not only the TLR4 pathway but also multiple inflammatory mediators, and that this combined suppression leads to the marked anti-inflammatory effect of DHP-3.
3) Postulated molecular mechanism underlying DHP-3-mediated suppression of the TLR4 pathway
Anti-inflammatory chemicals can suppress the TLR4 pathway via several mechanisms, such as activation of negative feedback, antagonism of TLR4 through direct binding, and enhancement of TLR4 degradation. We assessed the function of DHP-3 as an enhancer of negative feedback. We focused on MAPK phosphatase-1 (MKP-1), which regulates phosphorylation levels of MAPKs (ERK, JNK, and p38). DHP-3 significantly upregulated MKP-1 protein levels in LPS- stimulated cells, which was consistent with DHP-3 reducing levels of phosphorylated MAPKs.
We also examined ATF3, a repressor involved in negative feedback of the TLR4 pathway.
DHP-3 upregulated ATF3 protein levels in LPS-stimulated cells. These results indicated that DHP-3 enhanced negative feedback of the TLR4 pathway, which resulted in significant suppression of inflammation.
In the present study, we demonstrated that the anti-inflammation effect of DHP-3 results from suppression of the TLR4 pathway. The TLR4 pathway is involved in the development and pathology of numerous diseases, but there is no drug that directly targets the pathway. We propose that DHP-3 can suppress inflammation although glycation products are generally believed to be toxic for health. Further studies are necessary to investigate the effects of DHP-3 especially in vivo. We suggest that DHP-3 has the potential to be used for several clinical applications in the future.
本論文は学術雑誌に掲載された次の論文を基礎とするものである。
The Effect of Dihydropyrazines on Lipopolysaccharide-Stimulated Human Hepatoma HepG2 Cells via Regulating the TLR4-MyD88-Mediated NF-κB Signaling Pathway.
The Journal of Toxicological Sciences, 45, 401-409 (2020) Madoka Esaki, Takumi Ishida, Yuu Miyauchi and Shinji Takechi
本論文で使用した主な略語一覧表
AGEs advanced glycation end products AP-1 activator protein-1
ATF3 activating transcription factor 3 CCK-8 cell count kit-8
CCL2 CC-chemokine ligand 2 cDNA complementary DNA COX-1 cyclooxygenase-1 COX-2 cyclooxygenase-2
CREB cAMP response element binding protein DHP dihydropyrazine
DHP-3 3-hydro-2,2,5,6-tetramethylpyrazine DMSO dimethyl sulfoxide
ERK extracellular signal regulated protein kinase FBS fetal bovine serum
GAPDH glyceraldehyde-3-phosphate dehydrogenase IgG immunoglobulin G
IκB inhibitor of nuclear factor-κB IL interleukin
JNK c-Jun N-terminal kinase LDH lactate dehydrogenase LPS lipopolysaccharide
MAPK mitogen-activated protein kinase
MKP-1 mitogen-activated protein kinase phosphatase-1 mRNA messenger RNA
MyD88 myeloid differentiation primary response gene 88 NF-κB nuclear factor-κB
Nrf2 nuclear factor erythroid 2-related factor 2 PAGE polyacrylamide gel electrophoresis PBS phosphate buffered saline
PVDF polyvinylidene difluoride
q-PCR quantitative polymerase chain reaction RPMI Roswell Park memorial institute
SDS sodium dodecyl sulfate TLR4 Toll-like receptor 4
TNFα tumor necrosis factor alpha
目次
第1章 緒論
第1節 糖化産物………... 1
第2節 DHPs……….. 2
第2章 DHP-3処理によるTLR4経路シグナルの抑制 第1節 序………... 4
第2節 DHP-3処理による細胞生存試験および細胞毒性試験………... 6
第3節 DHP-3処理によるTLR4経路シグナル関連因子の発現抑制……….... 8
第1項 細胞膜受容体TLR4の発現抑制……… 8
第2項 アダプタータンパク質MyD88の発現抑制………... 11
第3項 NF-B経路関連因子の発現抑制………. 12
第4項 MAPK経路関連因子の発現抑制………. 19
第4節 考察………. 26
第5節 小括………. 28
第3章 DHP-3処理による炎症制御因子の産生抑制 第1節 序………. 29
第2節 炎症性サイトカイン転写抑制………. 29
第3節 ケモカイン転写抑制………. 32
第4節 エイコサノイド産生酵素COX-2の発現抑制………... 34
第5節 考察………. 36
第6節 小括………. 38
第4章 DHP-3処理によるTLR4経路抑制のメカニズム解明 第1節 序………. 39
第2節 ホスファターゼMKP-1の発現量変化………. 39
第3節 リプレッサーATF3の発現量変化………. 42
第4節 考察……….. 44
第5節 小括……….. 46
第5章 総括………. 47
実験の部………. 49
謝辞………. 55
参考文献………. 56
1 第1章 緒論
第1節 糖化産物
糖化産物は、グルコースなどの還元糖とアミノ基を有するタンパク質が非酵素的反応 によって縮合するメイラード反応により生じる化合物の総称であり、生体内に数十種類 存在すると言われている。1) メイラード反応は食品の加熱調理や長期保存する際に生じ る褐変反応として長年研究されてきた。近年、糖化産物は医療分野で糖尿病バイオマー カーとして、糖化ヘモグロビン (HbA1c) が用いられて、注目されている。2)
糖化産物の生成過程は前期および後期反応に分けることができる。還元糖(炭水化物)
とアミノ酸(タンパク質)が縮合し、アマドリ転位物が生成するまでの前期反応、次い で、酸化、脱水、縮合、転位反応によって不可逆的な構造変化が生じる後期反応に分か れ、それらの過程において様々な糖化産物類縁体が生成されている (Fig.1)。
数多くの糖化産物がある中で最も盛んに研究が行われている糖化産物は、終末糖化産 物 (advanced glycation end products; AGEs)である。AGEsは生体内タンパク質を修飾し、
タンパク発現の変化や酵素活性の低下などを引き起こす。AGEsは加齢と共に生体に蓄 Fig. 1. Maillard reaction
2
積され、加えて、糖尿病や糖尿病合併症、アテローム性動脈硬化症、非アルコール性脂 肪肝炎、神経変性疾患のアルツハイマーやパーキンソン病の発症などの様々な病態の発 症に関与している。 3-6)一方、近年一部のAGEs は、抗酸化作用や抗変異原性作用を有 し、ヘリコバクターピロリの除去においては、治療を促進する可能性も示唆されてい る。7-10)
生体内にはAGEsを含む糖化産物の生成経路が多数存在し、組織や病態によって存在 する糖化産物は異なる。したがって、糖化産物の作用について網羅的かつ包括的に解析 することは意義があり、引いては、超高齢化社会を迎えた我が国での加齢に伴い蓄積す る糖化産物の作用解明は喫緊の課題と考える。
第2節 DHPs
糖化産物dihydropyrazines (DHPs) は、生体内において、グルコサミンの2量体やアミ
ノレブリン酸の2量体より生成する。11, 12) S. Takechiらは、DHPsのラジカル産生能、酸 化ストレスの惹起、酸化型グルタチオンの増加、DNA 切断活性などの諸性質を解明し
ている。13-16) また最近の研究において、3-hydro-2,2,5,6-tetramethylpyrazine (DHP-3) (Fig.2)
は、nuclear factor erythroid 2-related factor 2 (Nrf2) 経路を活性化し、heme oxygenase-1や glutamate cysteine ligase catalytic subunitなどの酸化ストレス防御系遺伝子の発現を上昇 させることを明らかにした。17) Nrf2 は酸化ストレス防御のみならず、炎症制御におい ても重要な因子であることが分かっている。18, 19) そこで、本研究では、DHPによる炎 症制御の機構を検証することを主眼に、炎症反応に関与する細胞内シグナル経路の一つ であるToll-like receptor 4 (TLR4) 経路へDHP-3が与える影響を解析した。
第2章では、DHP-3がTLR4経路の各因子の発現レベルに及ぼす影響を検討、第3章
3
では、DHP-3による炎症制御因子の産生抑制に関する検討、第4章では、DHP-3による
TLR4経路抑制機構の解明を目指した。
以下に得られた知見を詳述する。
2
Fig. 2. (A) Chemical structure of 3-hydro-2,2,5,6-tetramethylpyrazine (DHP-3). (B) Synthesis of DHP-3 from 2,3-butanedione and 1,2-diamino-2-methylpropane.
(A)
(B)
4
第2章 DHP-3処理によるTLR4経路シグナルの抑制
第1節 序
Toll 様 受 容 体(TLR)は 、 様 々 な 病 原 体 を 認 識 す る パ タ ー ン 認 識 受 容 体 (pattern recognition receptor; PRR) の一つであり、T 細胞を活性化することで獲得免疫に寄与す る。20) 本研究では、TLR ファミリーの中で、グラム陰性細菌のリポポリサッカライド
(lipopolysaccharide; LPS)を認識し、炎症反応を引き起こすTLR4に着目し、DHP-3が 与える影響を精査した。21)
TLR4経路を介するシグナル伝達経路は、myeloid differentiation primary response gene
88 (MyD88) 依存的経路とMyD88非依存的経路の2種つに大別される。22) 本研究では、
特に炎症反応機構を誘導する MyD88 依存的経路に着目し、検討を行った (Fig.3) 。
MyD88は、TLR4アダプタータンパク質であり、TLR4がLPSなどのリガンドと結合す
ると、TLR4のToll/IL-1 receptorドメイン近傍に結合し、下流へシグナル伝達を行う。23) TLR4-MyD88 依存的経路の下流は nuclear factor-κB (NF-B) 経路と mitogen-activated protein kinase (MAPK) 経路により構成されている。TLR4へのLPS刺激は、NF-B経路 と MAPK 経路の両方を活性化し、炎症性サイトカイン産生を促すことで、炎症反応を 惹起する。24)
ヒトにおいて病原体による TLR4 経路活性化は、敗血症を引き起こす要因とされ、
致死率が非常に高い。25) 全世界の死者の5 人に1 人は敗血症により死亡していること が調査により明らかとなっているが、現時点では、敗血症に直接作用する治療薬はなく、
対症療法が治療の中心となっている。26) また、TLR4は感染症に関与しているだけでは なく、肝疾患やがんの転移などに関与しているとの報告もあり、シグナル伝達因子とし
5 ても注目されている。27, 28)
既に、ヒト肝がん由来HepG2細胞をDHP-3処理すると、Nrf2経路が活性化されるこ とが示唆されていることから、DHP-3は細胞に対し抗炎症作用を有することが想定され る。17)
本章では、DHP-3処理によるTLR4経路への影響を検討することにより、DHP-3の抗 炎症作用を精査した。実験には、ヒト肝がん由来株化細胞株 HepG2 細胞、ヒト前骨髄 性白血病由来株化細胞株HL60細胞、マウスマクロファージ由来株化細胞株J774.1細胞 を用いた。
Transcription Factors
MAPK pathway
NF-κBp50 NF-κBp65IκB
P
P ubiquitination
degradation
NF-κBpathway
MAPKKK
MAPKK
ERK JNK p38
AP-1
inflammation
TLR4 LPS
MyD88
nucleus
Fig. 3. TLR4 pathway
6
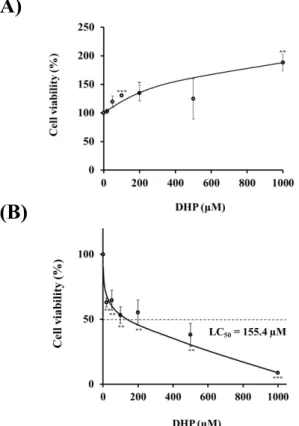
第2節 DHP-3処理による細胞生存試験および細胞毒性試験
本研究に先立ち、DHP-3 の細胞生存試験および細胞毒性試験を行い、本研究の
DHP-3処理濃度の範囲を決定した。
DHP-3の細胞生存試験はCell Counting Kit-8 (CCK-8)を用いて、生細胞の呼吸活性
を測定した。DHP-3の処理濃度を20-1000 µM に設定し、処理1時間後および24時
間後のHepG2細胞生存率を調べた。DHP-3 の1 時間処理は、細胞生存率の低下は確
認されなかった (Fig.4 A)。Fig.4 Bに示すように、DHP-3の24時間処理は濃度依存的 に、細胞生存率が低下した(LC50 = 155.4 µM)。
Fig. 4. Cell viability of DHP-3 in HepG2 cells. The cell viability was determined by CCK-8 assay after exposure to DHP-3 (20-1000 µM) for 1hr (A) and 24 hr (B). The control was culture medium containing dimethyl sulfoxide (DMSO). Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01, *** p < 0.001 indicate significant differences from the control group.)
(A)
(B)
7
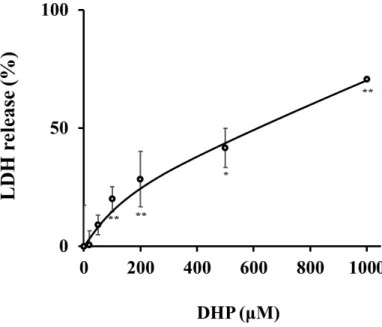
あわせて、傷害を受けた 細胞より培地中に放出された乳酸脱水素酵素(lactate dehydrogenase; LDH)活性を測定することにより細胞傷害を測定評価する Cytotoxicity
LDH Assay Kit-WSTを用い、細胞障害を評価した。細胞生存率の実験と同様に、DHP-3
処理24時間後に解析を行った。結果、DHP-3濃度依存的に、細胞からのLDHの放出量 が上昇し、細胞への障害が認められた (Fig.5)。
細胞生存試験およびLDL活性測定評価により、500 µM以上のDHP-3処理は細胞障
害により HepG2 細胞の半数以上を死に至らしめることを確認した。そこで、本研究の
DHP-3処理濃度を50-400 µM、処理時間を1時間に設定し、以降の解析を進めた。
なお、ヒトの DHP血中濃度が約 100 µMであることを考慮すると(当研究室未発表 データ)、今回用いたDHP-3濃度 (50-400 µM) は生理学的な範囲の濃度である。
Fig. 5. Cytotoxic effect of DHP-3 in HepG2 cells. LDH release was evaluated after exposure to DHP-3 (20-1000 µM) for 24 hr. The control was culture medium containing DMSO. Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01 indicate significant differences from the control group.)
8
第3節 DHP-3処理によるTLR4経路シグナル関連因子の発現抑制
第1項 細胞膜受容体TLR4の発現抑制
TLR4 は、LPS などのリガンドを認識することで、TLR4 経路の起点となり、下流の 様々な因子の発現や活性化を引き起こす。20) 通常 LPS で刺激した細胞は、TLR4 の転 写活性化やタンパク質発現上昇がみられる。まずは、DHP-3がTLR4の転写レベルに与 える影響をquantitative polymerase chain reaction (q-PCR)で解析した。LPS (1 µg/mL) で刺 激した HepG2 細胞は 200 µM の DHP-3 で処理することで、TLR4 の messenger RNA (mRNA) 発現量をLPS刺激細胞と比較して、46 ± 10%発現誘導を抑制した (Fig.6)。
HepG2
Fig. 6. DHP-3 inhibits transcription of TLR4 in LPS-stimulated HepG2 cells. The TLR4 mRNA levels were analyzed using q-PCR after normalization to glyceraldehyde-3- phosphate dehydrogenase (GAPDH) mRNA levels. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to 200 µM DHP-3 for 1 hr. Values represent the mean
± S.D. of 3 samples. (##p < 0.01 indicate significant differences from the untreated group.
**p < 0.01 indicate significant differences from the LPS-stimulation without DHP-3 group.)
9
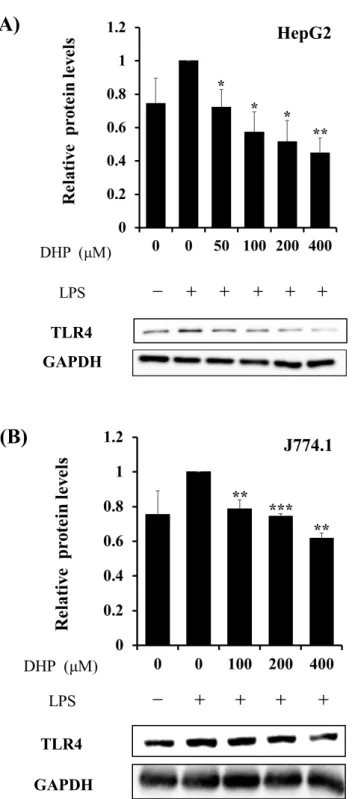
次いで、DHP-3処理がTLR4タンパク質発現量に与える影響を、ウエスタンブロッ
ティングを行った。LPSで刺激したHepG2細胞を各濃度に設定したDHP-3 (50, 100, 200,
400 µM)で処理することで、TLR4 のタンパク質発現量は LPS 刺激細胞と比較すると
55 ± 9%低下した (Fig.7 A)。また、免疫系細胞株のJ774.1細胞においても38 ± 3%低下
した (Fig.7 B)。
以上より、DHP-3は転写およびタンパク質レベルでTLR4の発現を抑制することが明 らかとなった。TLR4の発現低下は、LPSなどによる外部刺激を軽減することで、炎症 反応を抑制することが可能と期待される。
10
J774.1 TLR4
GAPDH
(A) HepG2
(B)
TLR4 GAPDH
Fig. 7. Effect of DHP-3 on protein expression of TLR4 in LPS-stimulated HepG2 cells (A) and J774.1 cells (B). Protein levels of TLR4 were detected by Western blotting. The band intensities of TLR4 were normalized to the expression levels of GAPDH. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 and J774.1 cells were exposed to various concentrations of DHP-3 (50-400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01,
***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group.)
11 第2項 アダプタータンパク質MyD88の発現抑制
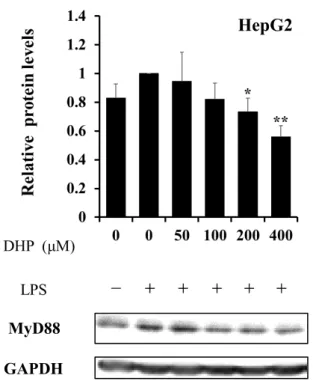
TLR4がLPSなどのリガンド刺激を受け活性化されると、TLR経路特有のアダプター タンパク質MyD88 を介し、下流に位置するNF-B経路や MAPK経路へシグナル伝達 が行われる。20) そこで、MyD88へDHP-3が与える影響を検討した。LPSでHepG2細 胞を刺激すると、MyD88 のタンパク質レベルは上昇傾向にあったが、各濃度の DHP-3 (50, 100, 200, 400 µM)で処理することで、この発現上昇が44 ± 8%抑制され、LPS未処理 レベルまでの低下を確認できた (Fig.8)。
MyD88 GAPDH
HepG2
Fig. 8. Effect of DHP-3 on protein expression of MyD88 in LPS-stimulated HepG2 cells.
After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. Protein levels of MyD88 were detected by Western blotting.
The band intensities of MyD88 were normalized to the expression levels of GAPDH.
Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01 indicate significant differences from the LPS-stimulation without DHP-3 group.)
12
第3項 NF-B経路関連因子の発現抑制
TLR4がLPSなどを認識すると、NF-B経路は活性化され、下流では、サイトカイン やケモカインなどの炎症反応を制御する因子の発現誘導が起きる。29) これらの機構に より細胞が炎症状態へと導かれる。不活性状態の NF-B は NF-B inhibitor である inhibitor of nuclear factor-κB (IB)と結合し、細胞質内に留まっている。TRL4が活性化さ れると、IB はリン酸化を受け、プロテアソームにより分解される。IB が解離した
NF-Bもリン酸化され、核内へ移行し、標的遺伝子の転写活性を促進する。
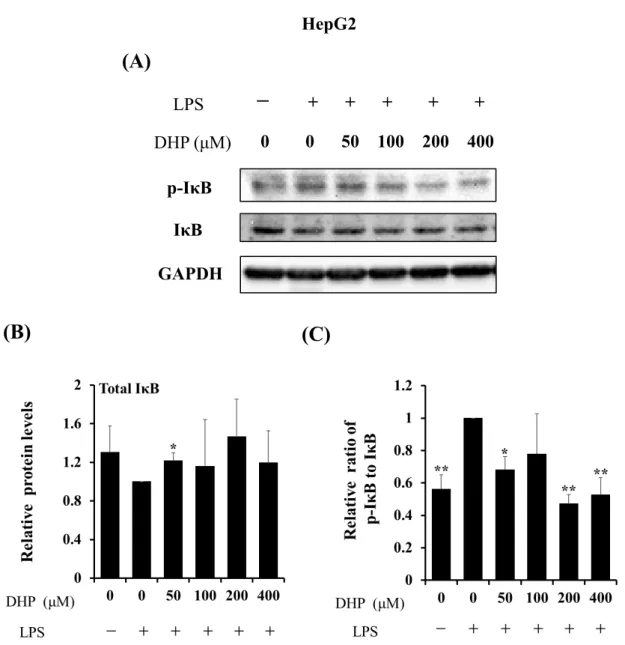
はじめに、LPSで刺激したHepG2細胞を、各濃度のDHP-3 (50, 100, 200, 400 µM)で処 理し、total IBおよびリン酸化 IB (Ser32/36)をウエスタンブロッティングでの解析を行 った結果、HepG2 細胞において、LPS 刺激により total IB の発現量は減少傾向が見ら れ、また、IBリン酸化レベルは上昇した (Fig.9 A and B)。細胞をさらにDHP-3で処理 することでtotal IBの発現量には増加傾向がみられ、リン酸化レベルは53 ± 6%低下し た (Fig.9 A and C)。
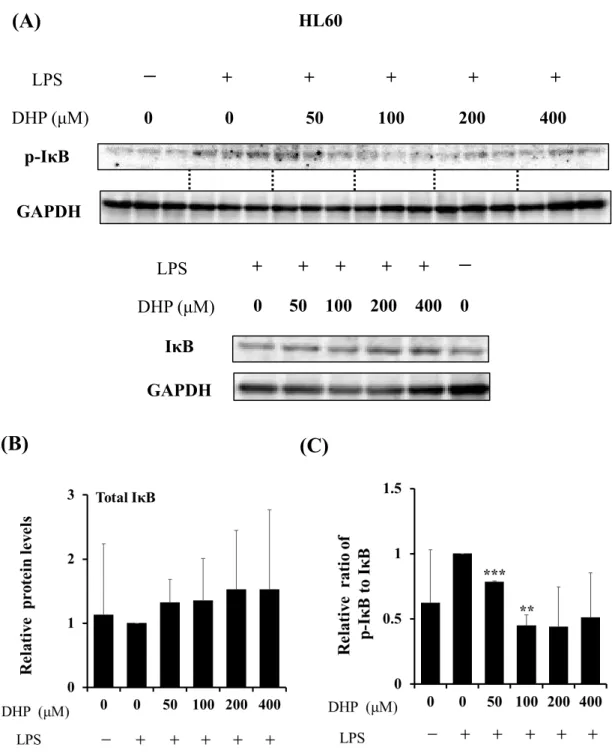
以上の結果から、DHP-3はLPS刺激によるIBのリン酸化を抑制することが示唆さ れた。また、HL60細胞を用いた検討においても同様の結果が得られた (Fig.10)。
13
IκB GAPDH
p-IκB
HepG2
(A)
0 0 50 100 200 400
-
+ + + + + LPSDHP (μM)
(B) (C)
Fig. 9. Effect of DHP-3 on activation of IκB in LPS-stimulated HepG2 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of IκB and p-IκB were examined by Western blotting. (B) The band intensities of total IκB were normalized to the expression levels of GAPDH. (C) The relative ratios of p-IκB/IκB were present. Values represent the mean ± S.D. of 3 samples.
(*p < 0.05, **p < 0.01 indicate significant differences from the LPS-stimulation without DHP-3 group.)
14
HL60
(A)
GAPDH p-IκB
0 0 50 100 200 400
-
+ + + + + LPSDHP (μM)
IκB
+ + + + +
-
LPS0 50 100 200 400 0 DHP (μM)
GAPDH
(B) (C)
Fig. 10. Effect of DHP-3 on activation of IκB in LPS-stimulated HL60 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HL60 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of IκB and p-IκB were examined by Western blotting. (B) The band intensities of total IκB were normalized to the expression levels of GAPDH. (C) The relative ratios of p-IκB/IκB were present. Values represent the mean ± S.D. of 3 samples.
(**p < 0.01, ***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group.)
15
DHP-3はIBのリン酸化および分解を抑制することが示唆されたため、DHP-3が
NF-Bへ与える影響を、HepG2細胞およびJ774.1 細胞を用いて解析した。NF-Bは、
受容体からのシグナルをClassical経路もしくはAlternative経路へ伝達する。30)
Classical経路は、サイトカインやケモカインなどの炎症反応に重要な因子の発現誘導
に関与する。Classical経路の NF-Bはp50/p65と2量体で挙動しており、本項では、
p65へDHP-3が与える影響に着目し精査した。
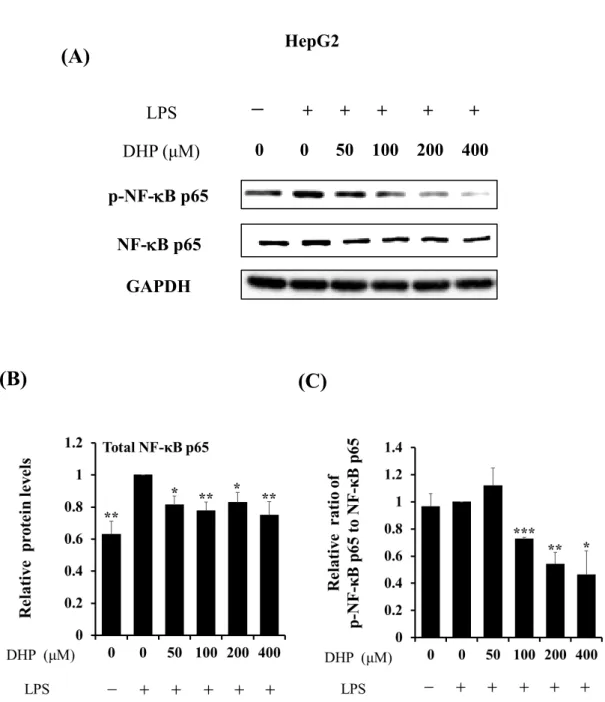
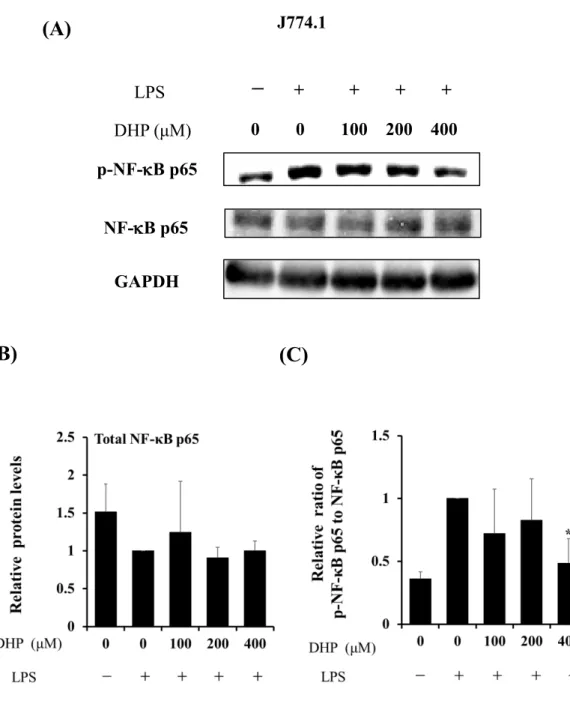
ウエスタンブロッティングにて、Total NF-B p65およびリン酸化 NF-B p65 (Ser536) を検出した。LPSで刺激したHepG2細胞をDHP-3処理すると、total NF-B p65の発現 量 の低下が 見られた が (Fig.11A and B)、 リン 酸化 レベ ルは 53 ± 18%低 下し た (Fig.11A and C)。あわせて、J774.1細胞を用いた検討においても、DHP-3処理により、
リン酸化 NF-B p65量はLPS刺激細胞と比較すると52 ± 20%低下した (Fig.12)。
16
(A)
0 0 50 100 200 400
-
+ + + + + LPSDHP (μM) p-NF-B p65
GAPDH NF-B p65
HepG2
(B) (C)
Fig. 11. Effect of DHP-3 on activation of NF-κB in LPS-stimulated HepG2 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of NF-κB p65 and p-NF-κB p65 were examined by Western blotting. (B) The band intensities of total NF-κB p65 were normalized to the expression levels of GAPDH. (C) The relative ratios of p-NF-κB p65/NF-κB p65 were present. Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01,
***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group.)
17
J774.1
(A)
0 0 100 200 400
-
+ + + + LPSDHP (μM) p-NF-B p65
GAPDH NF-B p65
(B) (C)
Fig. 12. Effect of DHP-3 on activation of the NF-κB in LPS-stimulated J774.1 cells. After 1 μg/mL LPS-stimulation for 6 hr, the J774.1 cells were exposed to various concentrations of DHP-3 (100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of NF-κB p65 and p-NF-κB p65 were examined by Western blotting. (B) The band intensities of total NF-κB p65 were normalized to the expression levels of GAPDH. (C) The relative ratios of p-NF-κB p65/NF-κB p65 were present. Values represent the mean ± S.D. of 3 samples. (*p < 0.05 indicate significant differences from the LPS-stimulation without DHP-3 group.)
18
DHP-3は、NF-B p65のリン酸化レベルを低下させることが明らかとなったため、次
に、DHP-3がNF-B p65の核内移行へ与える影響を検討した。細胞から核抽出を行い、
ウエスタンブロッティングにて解析した。LPS で刺激した HL60 細胞を、DHP-3
(200 µM) で処理し、細胞の核を抽出した。Fig. 13 に示すように、LPS刺激した細胞で
はNF-B p65が核内移行を顕著に示した。しかし、さらにDHP-3処理を行うと、細胞
の核内NF-B p65量は著しく低下した。これらの結果より、DHP-3処理がNF-B p65の
核内移行を抑制することが明らかとなった。また、DHP-3のみで処理した細胞において
は、NF-B p65の核内移行の増加などは観察されなかった。
Nuclear NF-κB p65 C23
HL60
Fig. 13. Effect of DHP-3 on nuclear translocation of NF-κB in LPS-stimulated HL60 cells.
After LPS-stimulation for 6 hr, the HL60 cells were exposed to 200 µM DHP-3 for 1 hr.
Extraction of the nuclear from HL60 cells was performed using EpiXtracTM Nuclear Protein Isolation Kit. The control was culture medium containing DMSO and LPS. The band intensities of NF-κB p65 were normalized to the expression levels of C23, a nuclear marker protein. Values represent the mean ± S.D. of 3 samples. (**p < 0.01, ***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group.)
19 第4項 MAPK経路関連因子の発現抑制
NF-B経路同様に、LPSなどをTLR4が認識することで、MAPK経路も活性化され、
炎症性サイトカインの産生を介し、細胞を炎症状態へ導く。MAPK 経路は細胞質内で は、最上流のMAPKKKからMAPKK、MAPKへとシグナル伝達が行われる。31, 32) 核内 において、これら上流シグナルを activator protein-1 (AP-1) が引継ぎ、標的遺伝子の 上流 (プロモーター)配列に結合し、転写活性を変化させ、炎症反応機構を制御して
いる。33-35) 本項では、細胞内シグナル伝達因子である MAPK の extracellular signal
regulated protein kinase (ERK)、c-Jun N-terminal kinase (JNK)、p38 および核内シグナル 伝達因子AP-1の一つであるJunに着目し、DHP-3処理がMAPK経路に与える影響を解 析した。
LPSで刺激したHepG2細胞を、さらにDHP-3 (50, 100, 200, 400 µM) で処理し、MAPK 経路シグナル伝達因子 (ERK、JNK、p38) の total タンパク質発現量および、それらの リン酸化レベルに与える影響を、ウエスタンブロッティングを用いて解析した。
ERKはtotal ERKおよびリン酸化 ERK (Tyr204)を解析した。HepG2細胞をLPSで刺激 することにより、total ERK 量に変化はなかったが、リン酸化 ERK のレベルは上昇し た。このリン酸化レベルの上昇は、DHP-3処理により有意に抑制され、LPS刺激細胞と 比較すると、53 ± 7%低下した (Fig.14)。
20
(A)
0 0 50 100 200 400
-
+ + + + + LPSDHP (μM) p-ERK ERK GAPDH
HepG2
(B) (C)
Fig. 14. Effect of DHP-3 on activation of ERK in LPS-stimulated HepG2 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of ERK and p-ERK were examined by Western blotting. (B) The band intensities of total ERK were normalized to the expression levels of GAPDH. (C) The relative ratios of p-ERK/ERK were present. Values represent the mean
± S.D. of 3 samples. (*p < 0.05, **p < 0.01 indicate significant differences from the LPS- stimulation without DHP-3 group.)
(C)
21
JNKはtotal JNKおよびリン酸化 JNK (Thr183またはTyr185)を解析した。HepG2細胞を LPSで刺激すると、リン酸化JNKが増加する傾向にあった。さらにDHP-3で処理する ことにより、LPS刺激細胞と比較すると、total JNKタンパク質発現量は1.9 ± 0.15倍上 昇し、リン酸化 JNKレベルは72 ± 3%低下した (Fig.15)。また、J774.1細胞を用いて同 様の検討を行ったところ、DHP-3処理によるtotal JNKタンパク発現量変化は確認され なかったが、リン酸化 JNKのタンパク発現量は55 ± 6%低下した (Fig.16)。
22
HepG2
(A)
0 0 50 100 200 400
-
+ + + + + LPSDHP (μM) p-JNK
GAPDH JNK
(B) (C)
Fig. 15. Effect of DHP-3 on activation of JNK in LPS-stimulated HepG2 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of JNK and p-JNK were examined by Western blotting.
(B) The band intensities of total JNK were normalized to the expression levels of GAPDH.
(C) The relative ratios of p-JNK/JNK and were present. Values represent the mean ± S.D.
of 3 samples. (*p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group.)
(C)
23
J774.1
(A)
0 0 100 200 400
-
+ + + + LPSDHP (μM) p-JNK
GAPDH JNK
(B) (C)
Fig. 16. Effect of DHP-3 on activation of JNK in LPS-stimulated J774.1 cells. After 1 μg/mL LPS-stimulation for 6 hr, the J774.1 cells were exposed to various concentrations of DHP-3 (100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of JNK and p-JNK were examined by Western blotting.
(B) The band intensities of total JNK were normalized to the expression levels of GAPDH.
(C) The relative ratios of p-JNK/JNK were present. Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01 indicate significant differences from the LPS-stimulation without DHP-3 group.)
(C)
24
p38はtotal p38およびリン酸化 p38 (Tyr182)を解析した。HepG2細胞をLPSで刺激す ることで、p38 のリン酸化レベルは大きく上昇した。一方、DHP-3 処理によりこの リン酸化レベルの上昇は有意に抑制され、LPS刺激細胞と比較すると、リン酸化 p38は 54 ± 3%低下した (Fig.17)。
(B) (C)
(A)
0 0 50 100 200 400
-
+ + + + + LPSDHP (μM) p-p38
GAPDH P38
HepG2
Fig. 17. Effect of DHP-3 on activation of p38 in LPS-stimulated HepG2 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. (A) Protein levels of p38 and p-p38 were examined by Western blotting.
(B) The band intensities of total p38 were normalized to the expression levels of GAPDH.
(C) The relative ratios of p-p38/p38 were present. Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group.)
(C)
25
上記のように、DHP-3処理はLPS刺激によるMAPKの活性化を抑制することが示唆 された。続いて、MAPK の下流にあり標的遺伝子の転写を制御する転写因子 AP-1 へ
DHP-3が与える影響について解析した。今回は、LPS刺激した HepG2細胞を、各濃度
のDHP-3 で処理し、AP-1の一つであるc-Junのタンパク質発現量を解析した。HepG2
細胞をLPSで刺激すると、c-Junの発現量は上昇した。この上昇は、さらにDHP-3処理 することで有意に抑制され、LPS 刺激細胞と比較すると、c-Jun のタンパク質発現量が 28 ± 2%低下した (Fig.18)。また、DHP-3のみで処理した細胞においても、LPS未処理細 胞と比較すると、c-Junのタンパク質発現量が29 ± 7%低下した。
HepG2
GAPDH c-jun
Fig. 18. Effect of DHP-3 on protein expression of c-Jun in LPS-stimulated HepG2 cells.
After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. Protein levels of c-Jun were detected by Western blotting. The band intensities of c-Jun were normalized to the expression levels of GAPDH. Values represent the mean ± S.D. of 3 samples. (*p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences from the LPS-stimulation without DHP-3 group. #p < 0.05 indicate significant differences from the untreated group.)
26 第4節 考察
本章では、DHP-3処理によるTLR4経路シグナルに対する抑制効果を検討した。結果、
DHP-3処理は複数の細胞種において、一様にTLR4の発現を低下させ、下流のNF-B経
路およびMAPK経路のリン酸化レベルを低下させることが明らかとなった (Fig.19)。
DHP-3 は細胞を炎症状態へ導くための炎症制御因子の産生に携わっている TLR4
経路全体の活性を抑制し、複数の炎症制御因子の産生を抑制する可能性が示唆され
た。36) DHP-3はTLR4経路をはじめとする炎症反応機構へ影響を与え、様々な炎症疾患
に治療的に作用することが期待できる。
DHP-3 は TLR4 経路シグナル関連因子を広範に抑制していることが明らかとなり、
DHP-3が示す本抑制のメカニズム候補は複数存在すると考えられる。まずは、DHP-3が
細胞外の因子である細胞膜受容体TLR4に直接影響を与え、TLR4経路シグナルを抑制 している可能性である。実際に TLR4 経路を抑制し抗炎症を発揮する化合物の中には、
TLR4 にリガンドとして直接結合しアンタゴニストとして働くものや、TLR4 を直接分 解するものが存在すると報告されている。21, 37, 38) DHP-3処理によりTLR4のタンパク質 発現量低下が確認されているため、DHP-3はTLR4のアンタゴニストもしくは分解促進 因子として働いている可能性がある。
別のメカニズムとしては、DHP-3 が細胞内で直接作用する可能性が挙げられる。
DHP-3が細胞内で起こしうる影響としては、DHP-3処理により複数のTLR4経路シグナ
ル関連因子のリン酸化レベルが一様に低下しているため、DHP-3がリン酸化酵素の活性 を低下させる、もしくは脱リン酸化酵素の活性を上昇させる可能性が考えられる。39)
DHP-3処理によるTLR4経路の抑制は、上記のようなメカニズムが一つもしくは複数重
なり合うことにより引き起こされていると推察される。
さらに、本章ではDHP-3がNF-B経路およびMAPK経路を共に抑制することを示唆
27
する結果が得られた。両経路の抑制を示唆させるものとして、total JNK発現上昇が考え られた。通常、NF-B経路とJNK経路はリンクしており、JNKのシグナル伝達経路は
NF-B 経路より抑制作用を受けている。40-42) しかしながら、今回、DHP-3 処理の影響
によりNF-B経路の活性化が抑制され、JNKシグナル伝達がNF-B経路より受けてい
た抑制系が解除されたことにより、total JNK が発現上昇を示したと推察される。そし て、通常、NF-B経路のJNK経路に対する抑制作用が解除されると、JNKのリン酸化 レベルは上昇がみられるが、DHP-3はリン酸化JNK の発現量を低下させた。このこと
は、DHP-3がTLR4 下流の複数のシグナル経路を抑制することを示唆するものである。
以上の知見より、DHP-3 は様々な TLR4 経路シグナル伝達因子の発現を低下させ、
LPS刺激によるTLR4経路活性化を抑制するものと考えられる。
Transcription Factors
MAPK pathway
NF-κBp50 NF-κBp65IκB
P
P ubiquitination
degradation
NF-κBpathway
MAPKKK
MAPKK
p38 ERK JNK
AP-1
inflammation
TLR4 LPS
MyD88
nucleus
Fig. 19. DHP-3 inhibits TLR4 signaling pathway.
28 第5節 小括
本章では、DHP-3 の炎症反応機構への影響を解析目的として、LPS 刺激細胞への
DHP-3処理よるTLR4経路の抑制効果について検討した。以下に、得られた知見を小括
する。
1) DHP-3は、TLR4の転写およびタンパク質発現量を低下させ、LPSなどによる外部
刺激を軽減することを明らかにした。
2) DHP-3は、TLR4アダプタータンパク質MyD88のタンパク質発現量を低下させ、
TLR4からのシグナルが、下流のNF-B経路やMAPK経路へ十分に伝達されない ことを確認した。
3) DHP-3 は、IB および NF-B のリン酸化レベルを低下させ、NF-Bの核内移行を
妨げ、NF-B経路の活性化を抑制することが判明した。
4) DHP-3は、3種のMAPK (ERK、JNK、p38) のリン酸化レベルを低下させ、c-Junの タンパク質発現の低下を引き起こし、MAPK 経路の活性化を抑制することが明ら かとなった。
29
第3章 DHP-3処理による炎症制御因子の産生抑制
第1節 序
生体内には、炎症反応を制御する因子が多数存在しており、サイトカイン、ケモカイ ンおよび脂質メディエーターなどが主となり働く。しかし、これらの炎症制御因子が過 剰に生体内で産生されると、好中球の異常活性化、血液凝固機能の活性化、血管拡張な どが生じ、敗血症性ショックや多臓器不全により、人を死に至らしめる。43, 44) また、こ のような炎症制御因子の過剰産生は、細菌やウイルスなどの病原体により生じるサイト カインストームの要因となっており、その直接的な治療方法の開発が急がれる。45, 46)
第2 章にて、DHP-3はLPS 刺激後の炎症反応機構TLR4 経路活性を抑制することが明
らかとなったため、本章では、DHP-3処理がTLR4経路の下流に位置する炎症制御因子 の産生に及ぼす影響を検討した。
第2節 炎症性サイトカイン転写抑制
サイトカインは様々な体細胞より産生されるポリペプチド性の生理活性物質であり、
炎症や免疫応答などの生体内にとっての緊急時に誘導され、生体防御因子として働 く。47) その一方で、炎症性サイトカインは、肝炎、関節リウマチ、アレルギー性疾患な どの病態形成に関与する。48-50) 本研究では、サイトカインの中でも、このような病態形 成に関与し、TLR4経路により制御を受ける炎症性サイトカインにDHP-3が与える影響 を解析した。
はじめに、DHP-3が代表的な炎症性サイトカイン、腫瘍壊死因子tumor necrosis factor
30
alpha (TNF)、interleukin-1beta (IL-1)、IL-6の転写活性に与える影響をq-PCRを用いて 解析した。51-53) HepG2細胞をLPSで刺激することより、TNF-、IL-1、L-6のmRNA 発現量は一様に上昇した。そして、このmRNA発現量の上昇は下記のように200 µMの
DHP-3処理により有意に抑制された。
TNF のmRNA発現量はLPS 刺激細胞と比較し、66 ± 12%抑制された (Fig.20 A)。 また、DHP-3とLPSの処理を入れ替えた場合においても63 ± 8%抑制された (Fig.20 A)。 IL-1 の mRNA 発現量は LPS 刺激細胞と比較し、45 ± 9%抑制された (Fig.20 B)。 また、DHP-3とLPSの処理を入れ替えた場合においても53 ± 24%抑制された (Fig.20 B)。
IL-6 の mRNA 発現量は LPS 刺激細胞と比較し、95 ± 4%と大きく抑制され た (Fig.20 C)。また、DHP-3とLPSの処理を入れ替えた場合においても92 ± 7%抑制さ れた (Fig.20 C)。
以上の結果より、DHP-3はLPSで刺激するタイミングに関わらずHepG2細胞からの 炎症性サイトカイン産生を転写レベルで抑制することが明らかとなった。
31
(A)
(B)
HepG2
Fig. 20. DHP-3 inhibits production of pro-inflammatory cytokines in LPS-stimulated HepG2 cells. The mRNA levels of TNF(A) L-1(B), and IL-6(C) were analyzed using q-PCR after normalization to GAPDH mRNA. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to 200 µM DHP-3 for 1 hr (pretreatment LPS). After 200 µM DHP-3 exposure for 1 hr, the HepG2 cells were stimulated by 1 μg/mL LPS for 6 hr (posttreatment LPS). Values represent the mean ± S.D. of 3 samples. (#p < 0.05 ##p < 0.01 indicate significant differences from the untreated group. **p < 0.01 indicate significant differences from the LPS-stimulation without DHP-3 group.)
(C)
32 第3節 ケモカイン転写抑制
生体内での炎症発症時には、白血球が組織に浸潤し、その制御をするのがケモカイン である。54) ケモカインは炎症反応だけではなく、炎症性疾患の増悪やがんの転移など にも関与する。55, 56) 本節では、DHP-3がケモカインの転写活性に与える影響をmRNA レベルで解析した。
炎症分野において他のケモカインと比較すると強力なヒスタミン遊離作用を 有するなど、炎症反応機構へ大きく関与するケモカインであり、かつ、糖化産物と関連 がある肥満やアテローム性動脈硬化症にも関与しているCC-chemokine ligand 2 (CCL2) を標的とし、DHP-3がその転写活性に与える影響を精査した。57-59)
LPS刺激でHepG2細胞を刺激すると、CCL2のmRNA発現量は上昇したが、 DHP-3
処理することで、CCL2 のmRNA 発現量は LPS 刺激細胞と比較し、84 ± 2%抑制され た (Fig.21)。また、DHP-3とLPSの処理を入れ替えた場合も、CCL2のmRNA発現量は LPS刺激細胞と比較して87 ± 10%抑制された (Fig.21)。
この結果より、DHP-3はLPS刺激条件に関わらずHepG2細胞からのケモカイン産生 を転写レベルで抑制することが示唆された。
33
HepG2
Fig. 21. DHP-3 inhibits production of chemokine in LPS-stimulated HepG2 cells. The mRNA levels of CCL2 were analyzed using q-PCR after normalization to GAPDH mRNA.
After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to 200 µM DHP-3 for 1 hr (pretreatment LPS). After 200 µM DHP-3 exposure for 1 hr, the HepG2 cells were stimulated by 1 μg/mL LPS for 6 hr (posttreatment LPS). Values represent the mean ± S.D.
of 3 samples. (#p < 0.05 indicate significant differences from the untreated group.*p < 0.05 indicate significant differences from the LPS-stimulation without DHP-3 group.)
34
第4節 エイコサノイド産生酵素COX-2の発現抑制
脂質は生体膜成分やエネルギーとして使用されるだけではなく、炎症制御因子や シグナル伝達分子としても働く。60, 61) 細胞膜リン脂質より遊離したアラキドン酸は cyclooxygenase-1/2 (COX-1, COX-2) を介し、炎症反応に関わるプロスタグランジンや トロンボキサンを生成する。62) COX-1は組織に恒常的に存在し、COX-2は炎症反応に 伴って誘導される。63)
本節では、DHP-3がCOX-2の発現に与える影響を検討した。LPSで刺激したHepG2 細胞をDHP-3 (50, 100, 200, 400 µM) で処理すると、COX-2のタンパク質発現量はLPS 刺激細胞と比較して最大で 34 ± 14%低下した。この低下は DHP-3 の処理濃度に依存 する傾向にあった (Fig.22)。また、DHP 単独処理した細胞においては、COX-2 の発現 上昇は観察されなかった。
これらの結果より、DHP-3はTLR4下流に位置する主な炎症制御因子の産生を抑制す ることで、抗炎症作用を示すことが示唆された。
35
HepG2
GAPDH COX-2
Fig. 22. Effect of DHP-3 on eicosanoid biosynthesis pathway in LPS-stimulated HepG2 cells. After 1 μg/mL LPS-stimulation for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was culture medium containing DMSO and LPS. Protein levels of COX-2 were detected by Western blotting.
The band intensities of COX-2 were normalized to the expression levels of GAPDH. Values represent the mean ± S.D. of 3 samples. (*p < 0.05 indicate significant differences from the LPS-stimulation without DHP-3 group.)
36 第5節 考察
本章では、DHP-3処理による炎症制御因子に対する産生抑制効果を検討した。結果と
しては、DHP-3処理はTLR4経路の活性化を抑制するのみならず、TLR4経路より産生
される炎症制御因子の生産まで抑制することが明らかとなり、DHP-3による抗炎症効果 がさらに明らかとなった。DHP-3は、炎症性サイトカイン、ケモカインおよびエイコサ ノイド産生酵素の産生を抑制した。複数の炎症性サイトカインを観察した中でも、IL-6 の転写活性が著しく抑制された要因としては、IL-6 の産生には NF-B の活性化が深く 関与しており、DHP-3がその活性化を抑制したため、IL-6の転写活性抑制がされたもの と考察する。64, 65)
炎症制御因子は白血球の遊走や炎症反応の構築へ大きく関与しているだけではなく、
TLR4経路のポジティブフィードバックとして働き炎症反応機構をさらに活性化させる。
TNF-、IL-1、L-6、CCL2 は産生されると、NF-B 経路および MAPK 経路をさらに 活性化状態へ導くことが報告されている。66-70) そのため、DHP-3 処理による TNF-、
IL-1、L-6、CCL2 の産生抑制は、第 2 章で述べた NF-B 経路および MAPK 経路の
抑制に関与していると推察する。
本章により DHP-3処理は TLR4 経路に対する抑制に加え、炎症制御因子の産生抑制 をすることが明らかとなったため、DHP-3がTLR4経路を抑制するメカニズム候補がさ らに一つ挙げられる。それは、TLR4 経路活性化をネガティブフィードバックする リプレッサーactivating transcription factor 3 (ATF3) が、DHP-3が引き起こす抑制機構に 関与する可能性である。71) ATF3は、TLR4経路のNF-B経路およびMAPK経路の抑制 作用に加え、炎症制御因子の産生を抑制する。72-74) DHP-3が炎症状態の細胞に生じさせ ている抑制機構はATF3による影響と類似しているため、DHP-3はATF3へ働きかけて