ケモインフォマティクスや創薬における
機械学習の世界初の本
Lodhi, H. and Yamanishi, Y.,
Chemoinformatics and Advanced Machine Learning Perspectives, IGI Global, 2010.
創薬の現状
費用も時間もかかる。
• 平均1000億円
• 10年以上
ゼロからの創薬は、ほとんど失敗する。
• 成功率は3万分の1
• 体内動態が不十分
• 毒性などの問題が途中で判明
ドラッグリポジショニング(
DR)
薬
再配置する(違う病気に)
n既承認薬や過去に医薬品開発に失敗した
化合物の新しい効能を発見し、別の疾患の
治療薬として開発
nヒトでの安全性や体内動態が確認され、製
造法も確立
n高速・低コスト・低リスク
医薬品開発の流れ
1. 創薬標的の同定 2. 大規模化合物スクリーニング 3. 化学構造最適化 4. 薬物動態試験 5. 非臨床試験 6. 第1相臨床試験(安全性の検証) 7. 第2、3相臨床試験(効果の検証) 8. 申請 従来 (10〜17年) ドラッグリポジショニング (3〜12年) 1. 創薬標的の同定 2. 既存薬スクリーニング 3. 化合構造最適化 4. 薬物動態試験 5. 非臨床試験 6. 第1相臨床試験(安全性の検証) 7. 第2、3相臨床試験(効果の検証) 8. 申請スキップ
(Nature Reviews Drug Discovery, 3, 673-683, 2004)
実例
nシルデナフィル(バイアグラ)
¨狭心症治療薬
→ 男性機能障害の薬
→ 肺高血圧症の薬
nミノキシジル(リアップ、ロゲイン)
¨高血圧治療薬
→ 発毛薬
より多くの患者さんを救済できる!
問題:これまでは偶然の発見に大きく依存していた
本研究では医薬ビッグデータに基づく
ドラッグリポジショニングを提案
オブジェクト データの例 薬物 薬理資料、臨床情報、化学構造、副作用報告、 治療標的タンパク質、オフターゲット、薬物応答 遺伝子発現情報、既知の効能など 低分子化合物 化学構造、標的タンパク質、生理活性情報など 遺伝子 タンパク質 アミノ酸配列、3次元立体構造、機能モチーフ、 パスウェイ、タンパク質間相互作用、分子機能、 生理学的役割、病理学的役割など 疾患 臨床情報、レセプト、電子カルテ、病因遺伝子、 環境因子、バイオマーカー、合併症情報、患者 の遺伝子発現情報、異常パスウェイなど薬と病気の関連を予測する機械学習法
(人工知能の基盤技術)を開発
薬1 病気1 病気2 病気3 すでに知られている効果 コンピュータ上で予測する新しい効果 薬2 薬3 薬4セレンディピティから脱却したい!
f (x, y) = w
Tφ
(x, y)
DRのためのAI創薬
多様な疾患の分子レベル
での理解が進んできた
n 病因遺伝子 n 発現異常遺伝子 n 疾患関連パスウェイ n 診断マーカー n 環境因子共通する
分子的特徴
疾患
A
疾患
B
遺伝子1 遺伝子2 遺伝子3提案手法の目的
様々な医薬ビッグデータから薬物・タンパク質・
疾患ネットワークを大規模予測する
x1 x2 x3 y1 y2 y3 薬(8千個) 疾患(2千個) 既知の関連エッジ 未知の関連エッジ (本研究で予測する) タンパク質(3万個) z1 z2 z3提案手法の目的
様々な医薬ビッグデータから薬物・タンパク質・
疾患ネットワークを大規模予測する
x1 x2 x3 y1 y2 y3 薬(8千個) 疾患(2千個) 既知の関連エッジ 未知の関連エッジ (本研究で予測する) タンパク質(3万個) z1 z2 z3 既承認薬でも、薬効に関与する標的タンパク質が不明なものは多い(約6割)タンパク質 etc. 薬物・化合物の化学構造 etc. ゲノム空間 ケミカル 空間
ケモゲノミクス法
フェノミクス法
人体へのフェノタイプ 薬理 空間 頭痛、吐き気、気分高揚、血圧の変化、疾患マーカーの変動、 etc.Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins Drugs Drugs A C B 転写 空間 薬物応答遺伝子発現
トランスクリプトミクス法
ゲノムワイドな薬物・タンパク質相互作用予測
ケモゲノミクスの手法
方針:化学構造が似ている薬は同様のタンパク質に
相互作用すると予測
(Yamanishi et al, Bioinformatics, 2008; Faulon et al., Bioinformatics, 2008; Jacob et al, Bioinformatics, 2008, Keiser et al, Nature, 2009; Yabuuchi et al, Mol Sys Bio, 2011)
タンパク質のアミノ酸配列・ドメイン・ モチーフの類似度
possible chemical substructures
薬の化学構造の類似度 475,692 KCF-S substructures (Kotera et al, BMC Syst. Biol., 2013)
kx(xi, xj) for i, j = 1, 2,..., nx kz(zi, zj) for i, j = 1, 2,..., nz 2012年度「理論分子生物学」講義予定表 ! ゲノム解析、ポスト・ゲノム解析とバイオインフォマティクス ! 配列アライメント、ダイナミックプログラミング法 ! ホモロジー検索、FASTA、BLASTアルゴリズム ! マルチプルアライメント、系統樹解析 ! 配列モチーフ ! 二次構造予測、膜貫通部位予測、立体構造予測 ! 遺伝子の機能アノテーション、比較ゲノム解析 ! ネットワーク解析 ! 分子生物学データベース ! 演習 ! http://goto.kuicr.kyoto-u.ac.jp/lecture/bioinfo.html 1 配列アライメント ! 配列アライメント(sequence alignment) ! 2つのタンパク質または遺伝子の配列を並べて、 進化的な関連があるかどうかを調べること ! 2つの遺伝子が進化的に関連があるか? ! 異なる生物種間で同じ機能を持つ遺伝子 ! 一つの生物種内で類似した機能を持つ遺伝子 真正細菌 古細菌 真核生物 原生生物 植物 菌類 動物 分子レベル(配列レベル)の情報:16S rRNA
生物種の系統関係
a a1 a2 遺伝子重複 a1 a2 a1 a2 種分岐 ホモログ(Homolog) 進化的な起源を同じくする遺伝子 オーソログ(Ortholog) 種分岐の際に同じ遺伝子だったもの 通常同じ機能を持つ パラログ(Paralog) 遺伝子重複によってできた類似遺伝子 通常異なる機能を持つ ゼノログ(Xenolog) 水平移動によって得られた類似遺伝子 a a1 a1 種分岐 種1 種2オーソログとパラログ
配列アライメント
! 配列アライメント ! 2つのタンパク質または遺伝子の配列を並べて、 ホモログ(相同)かどうかを調べること ! 実際には類似性を調べる ! 文字の一致(マッチ)、不一致(ミスマッチ)、 挿入、欠失を考慮する ! アライメントのキーポイントは ! アライメントの種類 ! アライメントの方法・アルゴリズム ! アライメントを評価するためのスコア ! スコアの重要性を評価するための統計的基準 ! グローバルアライメント ! 配列全体を並べる ! ローカルアライメント ! 局所的によく似た部分を探す LGPSSKQTGKGW-SRIWDN! | +| ||| |+ |! LN-ITKSAGKGAIMRLGDA! ---TGKG---! ||| ! ---AGKG---! マッチ(+, ¦) ミスマッチ ギャップ・挿入(-)配列アライメントの種類
Local sequence alighnment kernel
(Saigo et al, Bioinformatics, 2004)
Generalized Jaccard index
Drug space Feature space
Protein space
薬物・タンパク質相互作用の予測
A pairwise model for any drug-protein pair ( ʹx , ʹz ) :
f ( ʹx , ʹz ) = aijk((xi, zj), ( ʹx , ʹz )) j=1 nz
∑
i=1 nx∑
= aijkx(xi, ʹx )kz(zj, ʹz ) j=1 nz∑
i=1 nx∑
Step 1: Pairwise learning
Interacting pair Non-interacting pair Learning a model Drug similarity Protein similarity
薬物・タンパク質相互作用の予測
A pairwise model for any drug-protein pair ( ʹx , ʹz ) :
f ( ʹx , ʹz ) = aijk((xi, zj), ( ʹx , ʹz )) j=1 nz
∑
i=1 nx∑
= aijkx(xi, ʹx )kz(zj, ʹz ) j=1 nz∑
i=1 nx∑
Step 2: Predicting new interactions
Drug similarity Protein similarity Feature space Interacting pair Non-interacting pair Prediction New pairs
タンパク質 etc. 薬物・化合物の化学構造 etc. ゲノム空間 ケミカル 空間
ケモゲノミクス法
フェノミクス法
薬理 空間 頭痛、吐き気、気分高揚、血圧の変化、疾患マーカーの変動、 etc.Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins Drugs Drugs A C B 転写 空間 薬物応答遺伝子発現
トランスクリプトミクス法
人体へのフェノタイプゲノムワイドな薬物・タンパク質相互作用予測
トランスクリプトミクス法
方針:薬物応答遺伝子発現パターンが似ている薬(化
合物)は同様のタンパク質に相互作用すると予測
(Isker et al, Mol Syst Biol, 2013; Wong et al, Plos Comp Bio, 2013; Hizukuri et al, BMC Med Genomics, 2015; Iwata et al, Sci rep, 2017)
x = (x
1, x
2,
, x
22276)
T各薬物に対して、薬物応答の遺伝子発現比
(コントロールに対する)プロファイル
LINCS(16268薬物、77細胞、22276遺伝子)
化合物 の種類 細胞 の種類 druggene expression profile
高い閾値:類似構造の薬物が多いデータ 低い閾値:多様な構造の薬物だけのデータ
ベンチマークデータ(4
870個の相互作用)で
の性能評価:化学構造多様性を考慮
トランスクリプトミクス法の性能は化学構造に依存しない
ベンチマークデータの 化学構造多様性薬物の適応可能疾患の新規予測
n8270個の薬物(日本・欧米での既承認薬)に
対して、標的タンパク質(酵素、
GPCR、イオ
ンチャネルなど)を推定
n1325個の疾患(がん、神経変性疾患、免疫
系疾患、精神疾患など)に対する効能を予測
薬と疾患の
196048個の新しい関連を予測
(
6531薬と1132疾患)
ケモゲノミクスによる予測の例
n
ピオグリタゾン(
2型糖尿病の薬
)
¨
新しい適応疾患
: パーキンソン病
¨
推定されたタンパク質
: MAOB (monoamine oxidase B)
Pioglitazone
hydrochloride Similar compound in the learning set
(Quinn LP, et al. Br J Pharmacol., 2008)
トランスクリプトミクスによる予測例
n
フェノチアジン
(抗精神病薬)
¨
新しい適応疾患
: 前立腺がん
¨
推定タンパク質:
AR (androgen receptor)
Phenothiazine Similar compound in the
learning set
Enzalutamide
(Iwata et al, Sci Rep, 2017)
予測された
ARの阻害効果は
ウェット実験で確認できた
遺伝子発現変動から薬物が作用するパス
ウェイが分かる
Regulated genes Genes
In a pathway i k Not in a pathway r - i l - k
Total r l
n hypergeometric test
drug
gene expression profile
cell line down-regulated genes up-regulated genes 163 biological pathways in KEGG inactivated pathway activated pathway P-value
疾患
A
疾患
B
薬
(候補化合物)
既知の
治療効果
新しい
治療効果
分子機序の
類似性
疾患の類似性に基づくドラッグ
リポジショニング
化合物の毒性・副作用予測
候補化合物 etc. インシリコ予測 で代替できる? 人体への毒性 (数百種類) 心臓毒性、肝臓毒性、腎臓毒性、発癌性 etc.Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins
Drugs Drugs
A
C B
Targeted Proteins Drugs Side-effects
Molecular scale Phenotypic scale
Component 1 Component 2 Component 3 Targeted proteins Drugs Drugs A C B
• 医薬品・食品・化粧品開発では、化合物の効能だ
けでなく、毒性の適切な評価が重要
• 哺乳動物を用いた毒性試験は極めて高価(1件に
つき数千万円)で、世界的に哺乳動物を用いた毒
性試験が禁止される流れ
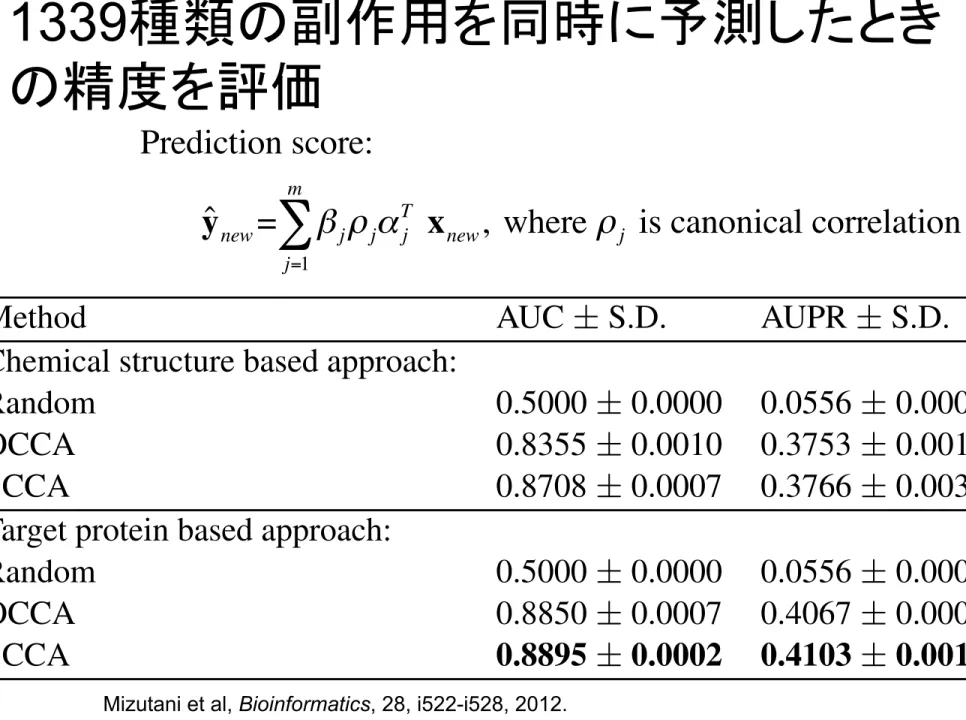
1339種類の副作用を同時に予測したとき
の精度を評価
Relating drug-protein interaction network with drug side-effects
Table 2. Most frequently appearing enriched pathways. ID KEGG pathway maps
map04080 Neuroactive ligand-receptor interaction map04020 Calcium signaling pathway
map04728 Dopaminergic synapse map04010 MAPK signaling pathway map04260 Cardiac muscle contraction map04727 GABAergic synapse
map04970 Salivary secretion map04725 Cholinergic synapse
map00590 Arachidonic acid metabolism
map04270 Vascular smooth muscle contraction
All component-based enriched pathways and associated enrichment scores are shown in Supplementary Table S2.
outcomes of biological pathway perturbations by drugs targeting the proteins that appear in the component.
4.3 Performance evaluation
It is difficult to evaluate the performance of the feature extraction method, because there is little knowledge about true association between targeted proteins and side-effects. However, if the extracted components are biologically meaningful, they should contain some general properties which could be exploited for side-effect prediction. We evaluate the performance of the method by recovering known drug side-effect profiles from drug protein binding profiles, using the extracted canonical components.
In previous literature, chemical structure fingerprints were used for predicting side-effect profiles in the framework of OCCA (Atias and Sharan, 2011) and SCCA (Pauwels et al., 2011). Therefore, we made a comparison between chemical structure-based approach and targeted protein-based approach in the framework of both OCCA and SCCA by performing the following 5-fold cross-validation. First, drugs in the gold standard set were split into five subsets of roughly equal sizes, and each subset was used in turn as a test set. Second, the CCA model was trained on the remaining four sets. Third, the prediction score was computed from the test set, based on the components extracted from the training set. Finally, the model was evaluated for prediction accuracy over the five folds.
We evaluated the performance of the methods by the ROC curve (receiver operating characteristic curve) and the Precision-Recall curve. The ROC curve is the plot of true positives as a function of false positives based on various prediction score thresholds, where true positives are correctly predicted side-effects and false positives are incorrectly predicted side-effects. The Precision-Recall curve is the plot of ”precision” (positive predictive value) as a function of ”recall” (sensitivity) based on various thresholds.
We summarized the performance by the AUC (area under the ROC curve) score and the AUPR (area under the Precision-Recall curve) score. To obtain robust results we repeated the cross-validation experiment five times, and computed the mean and the standard deviation (S.D.) of the AUC scores over the five repetitions.
Sparsity parameters c1, c2 ranged from 0 to 1 by 0.1 increments, and
m ranged from 10 to 200 by 10 increments. The best results were
obtained with c1 = 0.1, c2 = 0.1, and with m = 80 components
in the case of SCCA. The same cross-validation experiments were repeated for OCCA (with no sparsity constraint), and the best results were obtained for m = 20 components.
Table 3. Performance evaluation based on 5-fold cross-validation.
Method AUC ± S.D. AUPR ± S.D. Chemical structure based approach:
Random 0.5000 ± 0.0000 0.0556 ± 0.0000 OCCA 0.8355 ± 0.0010 0.3753 ± 0.0016 SCCA 0.8708 ± 0.0007 0.3766 ± 0.0030 Target protein based approach:
Random 0.5000 ± 0.0000 0.0556 ± 0.0000 OCCA 0.8850 ± 0.0007 0.4067 ± 0.0006 SCCA 0.8895 ± 0.0002 0.4103 ± 0.0018
Scores of the proposed method are highlighted in bold.
Table 3 shows the resulting AUC and AUPR scores for the four different approaches, where the prediction scores for all side-effects were merged, and a global ROC curve and a global PR curve were evaluated for each method. This indicates that both SCCA and OCCA produce fairly good results and SCCA is slightly better than OCCA. It also seems that the targeted protein-based approach works better than the chemical structure-based approach. Results suggest that the targeted protein information is indeed useful for side-effect prediction.
4.4 Prediction of side-effects for uncharacterized drugs
In the DrugBank database, there are still 730 drugs whose target protein information is available, but side-effects are not stored in the SIDER database. Based on their protein-binding profiles, we predicted the potential side-effects for these uncharacterized drugs using the SCCA model, all of the 658 reference drugs being used as a training set. All prediction results can be found in Supplementary Table S3(A). Complete analysis of all predictions is of course out of reach, so we focused on the side-effect predictions of highest scores. Some of the top ranked predicted side-effects involve Cinnarizine (DB00568), an anti-histaminic drug used against motion sickness. This drug binds to the Histaminic H1 receptor, which is believed to explain its indication against vomiting in motion sickness. Its predicted ”tremor” (cyclical movement of a body part) side-effect was confirmed by literature (Gimenez-Roldan and Mateo, 1991). Interestingly, Cinnarizine also binds to the voltage-dependent calcium channel involved in muscle contraction, which might explain the ”tremor” side effect. ”Constipation” is also predicted for Cinnarizine, as we could find in the adverse effect report 6127929-0 of the Food and Drug Administration (FDA). The predicted ”dry mouth” side-effect for Cinnazarine was also confirmed from literature (Gordon et al., 2001).
The second ranked predicted side-effect is ”diplopia” (double vision) for benzocaine (DB01086), a surface anesthetic that acts by preventing transmission of impulse along nerve fibers. Consistent with this activity, benzocaine is an inhibitor of voltage-dependent sodium channel. The predicted side-effect was confirmed from the literature (Horowitz et al., 2005). ”Syncope” was another side-effect predicted for benzocaine with a high score (Walker et al., 2003). The fourth ranked predicted side-effect is ”tremor” for Bepridil (DB01244), an antihypertensive drug. Tremor is indeed one of the most common side-effects for this drug, reported for 5 percent of all patients (Williams et al., 2002). ”Tachycardia” was also confirmed for Promazine (DB00420), an antipsychotic agent (Aronson, 2007). The side-effect ”diplopia” for Nisoldipine (DB00401), a calcium channel blocker used for the management of hypertension, was also confirmed (O’Keefe and Creamer, 1987).
5 Mizutani et al, Bioinformatics, 28, i522-i528, 2012.
Prediction score: ˆynew= βjρjαTj
j=1 m