Ethylene Oligo/Polymerization by Molecular Modeling
○Wenhong Yang1,2, Wen-Hua Sun11 Institute of Chemistry, Chinese Academy of Sciences, China 2 Department of Chemistry, School of Science, University of Tokyo, Japan
Introduction
In these decades, the characteristic catalytic activities of the late transition complex catalysts towards ethylene oligo/polymerization have been attracting the attention of researchers in the field catalytic chemistry [1]. Indeed, these catalysts have high stabilities as well as high catalytic activities. In addition, they can be synthesized at low cost. By the modification of substituents on ligands as well as by the introduction of alternative ligands, a variety of the late transition complex catalysts have been synthesized for optimizing the catalytic performances for specific applications [2]. However, it is still a difficult task to predict their catalytic performance by such modification of the ligands. The molecular modeling method can be used for investigating the catalytic reaction mechanism at the molecular level and can potentially be used for predicting the catalytic activities. In our recent studies, the catalytic activities of late transition metal complex catalysts were investigated on the basis of the electronic and steric effects using the multiple linear regressions analysis (MLRA) [3] and the two dimensional quantitative structure-activity relationship (2D-QSAR) modeling [4]. Furthermore, we investigated other catalytic performances such as molecular weight and melting temperature of the obtained polymerization products by 3D-QSPR modeling and showed that good predictive and validated capabilities.
Computational Methodology
The catalytic activities of transition metal complexes were investigated by the self-defined parameters using MLRA. Based on the optimized geometry obtained by DFT calculations, two parameters representing the electronic effect, that is, the effective net charge (𝑄) of the metal center and the Hammett constants (F), and two parameters representing the steric effect, that is, the open cone angle (θ) and the bite angle (β), were calculated, and the catalytic activity (A) is represented using these four parameters as
𝐴 = 𝑚0+ 𝑚1𝐹 + 𝑚2𝑄 + 𝑚3𝜃 + 𝑚4𝛽 (1)
For the big data set of the complexes, the catalytic activities were evaluated by the 2D-QSAR modeling method with the procedure shown in Scheme 1.
Results and Discussion
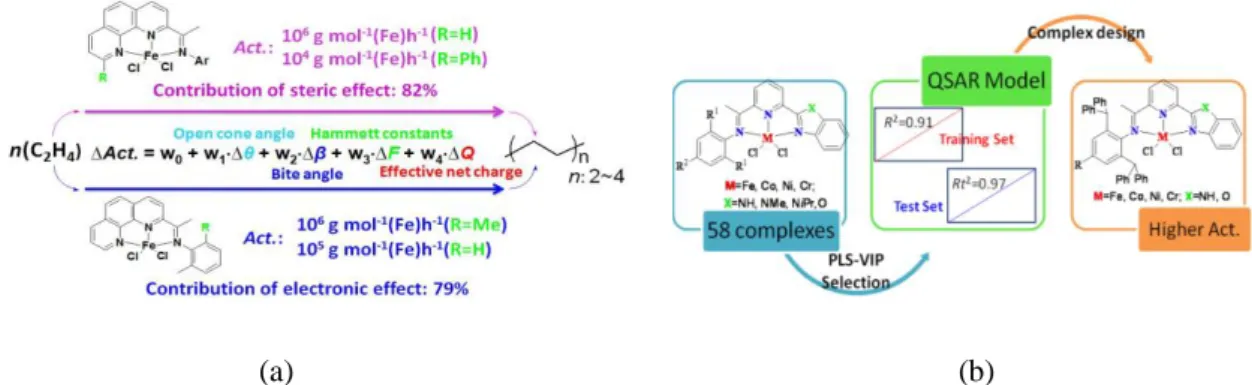
It has been known from experimental studies [3] that transition metal complexes systems ligated with the same ligands such as the series of 2-imino-1, 10-phenanthrolinylmetal complexes shown in Fig. 1a, exhibit a variety of different catalytic activities towards ethylene oligo/polymerization. Using the variation of the catalytic activities as a response variable and the change of the parameters as independent variables in the MLRA equation, we showed that the calculated catalytic activities exhibit a good correlation with the experimental results. The contributions in the electronic and steric effects were analyzed separately from each other to interpret the variations for their catalytic activities. It was found that the extent of the correlation among the analogues having different metal species is low compared with the extent of the correlation among the analogues having the same metal atom species.
The 2D-QSAR approach was adopted to investigate the catalytic activities for a data set of 58 2-azacyclyl-6-aryliminopyridylmetal complexes in Fig. 1b. Molecular descriptors were derived based on the optimized structure of complexes and were selected by the partial least square method (PLS). The final QSAR model containing 18 descriptors showed good predictive abilities for the training set of 36 complexes, for which the correlation coefficient value (R2) was 0.913 and the cross validation (Q2) was 0.873, and the test set of 16 complexes for which the correlation coefficient value (Rt2) was 0.971. Through the detailed analysis of the relationship between the selected descriptors and the cataltyic activites, 20 new complexes were designed and their catalytic activities were predicted. The present results are expected to give guidance for the design of transition metal complex catalysts with high catalytic activities.
(a) (b)
Figure 1. The investigation of the variation of the catalytic activities for the late transition metal complexes analogues by (a) the MLRA modeling method and (b) the 2D-QSAR modeling method.
References
[1]. Small, B.L.; Brookhart, M.; Bennett, A.M.A. J. Am. Chem. Soc. 1998, 120, 4049−4050. [2]. Flisak, Z.; Sun, W.-H. ACS Catal. 2015, 5, 4713−4724.
[3]. Yang, W.; Ma, Z.; Sun, W.-H. RSC Adv. 2016, 6, 79335−79342.
h-BN/Au (111)に担持した金クラスターの水素発生反応に対する
触媒活性に関する理論的研究
1北大院・総化,2北大院・理,3京大・ESICB,4NIMS GREEN,5JSTさきがけ ○中原真希1,高敏2,3,Andrey Lyalin4,小林正人2,3,5,武次徹也2,3,4
Hydrogen evolution reaction catalyzed by gold cluster
supported on h-BN/Au (111)
○Maki Nakahara1, Min Gao 2,3, Andrey Lyalin4, Masato Kobayashi2,3,5, Tetsuya Taketsugu2,3,4
1 Graduate School of Chemical Sciences and Engineering, Hokkaido Univ., Japan 2 Faculty of Science, Hokkaido Univ., Japan
3 ESICB, Kyoto Univ., Japan 4 NIMS GREEN, Japan 5 PRESTO JST, Japan
【Abstract】Hexagonal-boron nitride (h-BN) with doped atoms shows high catalytic activity, although h-BN is inert. Recently, it is reported that h-BN on gold substrates (h-BN/Au) has catalytic activity of oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER). Further, gold cluster supported on h-BN/Au was shown to be more active for ORR compared to h-BN/Au. The purpose of this study is to investigate the size effect of gold clusters on h-BN/Au for HER. The active site, charge effect, size effect of gold clusters supported on h-BN/Au have been studied. The results show that gold clusters of Aun-h-BN/Au were more active than the h-BN/Au surface. We suggest that electron transfer between gold clusters and h-BN/Au surface strongly influences the catalytic activity of supported gold clusters. The relationships between characteristics of structures of Aun-h-BN/Au and adsorption energy has also been investigated.
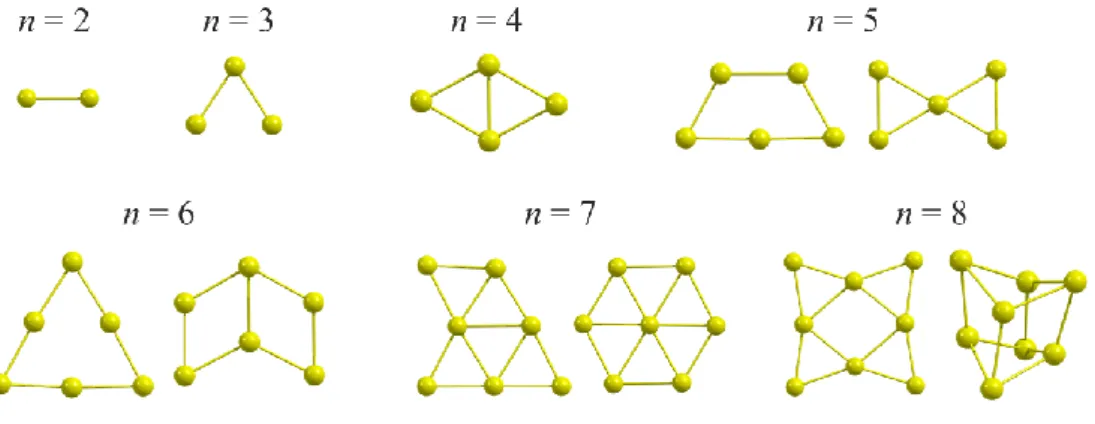
【序】 近年、hexagonal-Boron Nitride (h-BN) を用いた触媒に注目が集まっている。 h-BN 自身は不活性だが、欠陥の導入や他の材料と接合することによって高活性な触 媒として働く。最近の研究で、金表面に h-BN を乗せた触媒 (h-BN/Au) が酸素還元反 応 (ORR) と水素発生反応 (HER) に活性を示し、h-BN/Au の上にさらに金クラスタ ーを担持した触媒が ORR にさらに高い活性を示すことが報告された[1, 2]。本研究で はこの触媒に着目し、量子化学計算を用いて触媒の構造探索と水素発生反応の検討を 行った。また、機械学習を使用し、触媒の構造解析を行った結果も報告する。 【計算手法】 SIESTA を用いて DFT 計算を行った。h-BN/Au 表面のモデルは 7×7×4 の Au(111)に 8×8 の h-BN を乗せたものを使用し、周期境界条件を適用した。汎関数 には Wu-Cohen(PBE)を使用し、Mesh Cutoff を 100 Ry に設定した。水素発生反応に対 する触媒活性を評価するために、Volcano Curve 理論[3]を導入し、水素原子が触媒に 吸着する際の自由エネルギー変化 ∆GH*が 0 に近いものほど高活性な HER 触媒として 働くと仮定した。 【結果】 まず、n = 1~8 に対して報告されている Aunクラスターの安定構造(Figure 1) を使用し、h-BN/Au 上の様々な場所に Aunを乗せて構造最適化計算を行い、吸着エネ ルギーEadから Aun-h-BN/Au の最安定構造を探索した。Eadは Aunクラスターが h-BN/Au に吸着した状態の電子エネルギー(Eel)から両者が離れた状態の Eelを差し引くことに より算出した。Aunクラスターは h-BN/Au 上の窒素原子の近くで安定化されることが
示された。また、Aunクラスターは構造が平面的であり、h-BN/Au 表面に対し垂直に 立った状態で安定化される傾向があることがわかった。次に、上記の最安定構造を用 いて Aun-h-BN/Au 上の h-BN 表面や Aunクラスターに水素原子を吸着させて∆GH*を求 め、水素発生反応を検討した結果を示す(Figure 2)。∆GH*は水素原子が吸着した状態の Eelから離れた状態の Eelを差し引き、自由エネルギー変化を見積もるために 0.3 eV を 加えた。Figure 2 より、h-BN/Au 表面よりも担持した Aunクラスターが HER 触媒とし て働くことが示唆された。さらに、孤立した Aunクラスターと Aun-h-BN/Au の活性を 比較し、h-BN/Au 表面が活性に与える影響を調べた。その結果、Aun クラスターと h-BN/Au との間の電荷移動が触媒活性に影響を与えている可能性が示唆された。また、 Random Forest を用いて Aun-h-BN/Au の構造解析を行ったところ、触媒の構造と吸着 エネルギーとの間に相関があることが示された。詳細は当日報告する。
【参考文献】
[1] K. Uosaki et al., Sci. Rep., 2016, 6, 32217-6pages
[2] G. Elumalai et al., Electrochem, Commun., 2016, 66, 53-57 [3] S. Z. Qiao et al., Angew. Chem. Int. Ed., 2015, 54, 52-65
Figure 2 Free energy variations for HER on Aun-h-BN/Au Figure 1 Stable structures of Aun cluster (n = 1~8)
次世代有機太陽電池に関する自動材料探索スキーム
1首都大院理工, 2東洋大, 3理研AICS
○今村穣1,田代基慶2,河東田道夫3
Automatic Search Scheme for Next-generation Organic Solar Cells
○Yutaka Imamura1, Motomichi Tashiro2, Michio Katouda3
1 Graduate School of Science and Engineering, Tokyo Metropolitan University, Japan 2 Department of Applied Chemistry, Toyo University, Japan
3 Advanced Institute for Computational Science, RIKEN, Japan
【Abstract】Organic photovoltaic cells consisting of electron donor materials, usually
semiconducting polymers, and electron acceptor materials, usually fullerene derivatives, are one of promising solar cells since organic photovoltaic cells have good features of low cost and flexibility. However, organic photovoltaics has not reached the practical power conversion efficiency, 15% and, therefore, new organic photovoltaic materials should be developed. This study searches for novel organic photovoltaic materials by computational science. Specifically, we examine new donor-acceptor type polythiophene used as a donor material in organic photovoltaics.
Unit structures for the polythiophene are automatically generated using libraries for donor and acceptor units. Using frontier orbital levels, photovoltaic characteristics such as short-circuit current densities JSC, open-circuit voltage VOC, and power conversion efficiencies
(PCEs), were estimated. The results of photovoltaic characteristics are consistent with experimental results. 【序】次世代有機太陽電池である有機薄膜太陽電池は、低コストや低環境負荷を特長 とする新しいエネルギー源として注目されている。有機薄膜太陽電池では、電子供与 体と電子受容体から形成されている。これまで、電子供与体としてポリチオフェン誘 導体、電子受容体としてフラーレン誘導体が頻繁に用いられてきたが、最近ではすべ てポリマーから形成される有機薄膜太陽電池も開発されている。しかし、実用化の基 準である光電変換効率15%にはまだ達しておらず、今後更なる有機薄膜太陽電池材料 の開発・設計が必要である。本研究では、ライブラリから自動でポリマーを生成し、 太陽電池の特性を検討するスキームの開発を行っ た[1,2]。 【結果と議論】本研究では、ドナー・アクセプター 型の新規ポリチオフェン(図 1)に関して検討を行っ た。ドナー・アクセプター型ポリマーは、最高占有 軌道(HOMO)-最低非占有軌道(LUMO)のエネルギ ー準位の設計が容易であるため新規材料に最適で あり、実際に高い光電変換効率が報告されている。 有機半導体材料でよく用いられるドナーおよび アクセプター部位のライブラリを作成し、それに基 づきポリチオフェンの単位ユニットを自動生成し た。得られた軌道エネルギーから、太陽電池特性で 図1 ドナー・アクセプター型ポ リマーの軌道エネルギー 電子供与部位 (D) ポリマー基本単位 電子吸引部位 (A) ELUMO EHOMO ELUMO EHOMO EDIFF 軌道エネルギー D A
ある短絡電流(JSC)、開放電圧(VOC)、光電変換効率(PCE)を計算した。短絡電流は、以 下の式を用いて見積もった。
d EQE Jsc
( )*AirMass1.5( ) , (1)
d
EQE
(
)
0
.
65
*
(
GapPolymer)
, (2)Air Mass 1.5 の太陽光を用いた。EQE のパラメータは標準的な 0.65 とした。次に開放 電圧は、Scharber のモデルを用いて見積もった。
eV
3
.
0
Polymer HOMO Acceptor LUMO OC
eV
, (3) 電子受容体としてPCBM を仮定し、 Acceptor LUMO
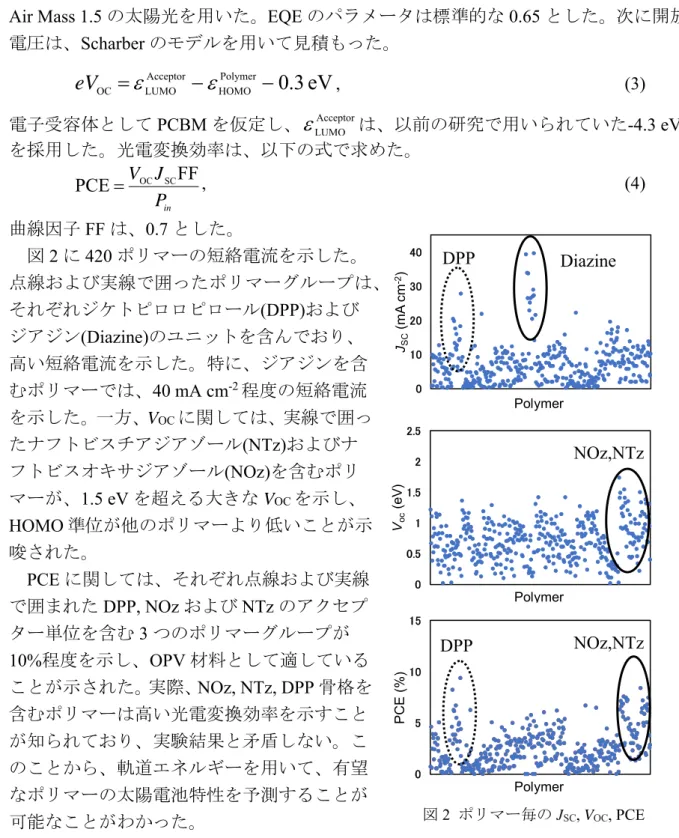
は、以前の研究で用いられていた-4.3 eV を採用した。光電変換効率は、以下の式で求めた。 in P J V FF PCE OC SC , (4) 曲線因子FF は、0.7 とした。 図2 に 420 ポリマーの短絡電流を示した。 点線および実線で囲ったポリマーグループは、 それぞれジケトピロロピロール(DPP)および ジアジン(Diazine)のユニットを含んでおり、 高い短絡電流を示した。特に、ジアジンを含 むポリマーでは、40 mA cm-2程度の短絡電流 を示した。一方、VOCに関しては、実線で囲っ たナフトビスチアジアゾール(NTz)およびナ フトビスオキサジアゾール(NOz)を含むポリ マーが、1.5 eV を超える大きな VOCを示し、 HOMO 準位が他のポリマーより低いことが示 唆された。 PCE に関しては、それぞれ点線および実線 で囲まれたDPP, NOz および NTz のアクセプ ター単位を含む3 つのポリマーグループが 10%程度を示し、OPV 材料として適している ことが示された。実際、NOz, NTz, DPP 骨格を 含むポリマーは高い光電変換効率を示すこと が知られており、実験結果と矛盾しない。こ のことから、軌道エネルギーを用いて、有望 なポリマーの太陽電池特性を予測することが 可能なことがわかった。[1] T. Matsui, Y. Imamura, I. Osaka, K. Takimiya, and T. Nakajima, J. Phys. Chem. C, 120, 8305 (2016) [2] Y. Imamura, M. Tashiro, M. Katouda, and M. Hada, J. Phys. Chem. C, to be submitted.
図2 ポリマー毎のJSC, VOC, PCE 0 10 20 30 40 JSC (mA cm -2) Polymer 0 0.5 1 1.5 2 2.5 Voc (e V ) Polymer 0 5 10 15 PC E (% ) Polymer DPP NOz,NTz NOz,NTz DPP Diazine
共有結合性有機構造体に包接されたキャリアの
イオン伝導機構の解明
1名大院・理, 2名大WPI-ITbM, 3北陸先端科学技術大学院大学, 4早大理工研, 5早大先
進理工, 6JST-CREST, 7京大ESICB, 8Oak Ridge National Laboratory
○林拓1, 土方優1,2, 江東林3, 西村好史4, 中井浩巳4,5,6,7, Stephan Irle8
Investigation of ion conductive mechanisms of carriers
in covalent organic frameworks
○Taku Hayashi1, Yuh Hijikata1,2, Donglin Jiang3, Yoshifumi Nishimura4, Hiromi Nakai4,5,6,7,
Stephan Irle8
1Grad. Sch. Sci., Nagoya Univ. 2WPI-ITbM, Nagoya Univ. 3JAIST 4RISE, Waseda Univ. 5Sch.
Advanced Sci. Eng., Waseda Univ. 6JST-CREST 7ESICB, Kyoto Univ. 8ORNL
【Abstract 】 Covalent organic frameworks (COFs) are crystalline porous polymers
consisting of organic molecules connected by covalent bonds. They can show ion conductivity when ion carriers are introduced into their pores and have been proposed as electrode-active materials in batteries and fuel cells. However, besides the lack of systematic design guidelines for COFs with high conductivity, the ion conduction mechanism and effects of the frameworks are still unclear. We therefore attempted to reveal the conduction mechanism using quantum mechanical molecular dynamics (MD) simulations. We employed the combination of density-functional tight-binding (DFTB) and divide-and-conquer (DC) methods, named DC-DFTB to perform long time simulations of large systems to analyze ion conduction mechanisms in a proton-conductive COF using imidazole as the ion carrier and an anion-conductive COF using water as the ion carrier. We will discuss the ion conduction mechanisms in the COFs, and effects of the frameworks on the mechanisms.
【序】 近年、電池や燃料電池の性能向上のため、高いイオン伝導性と熱的・化学的安定性 を兼ね備えた電解質の開発が求められている。最近では共有結合性有機構造体 (COF) を用いたイオン伝導系が注目されてきている。COF は有機分子が共有結合を介して形 成した結晶性多孔性物質であり、細孔内にイオンキャリア分子を導入することでイオ ン伝導性を示すという報告がなされている。COF は化学的安定性が高い上、骨格を構 成するユニットやゲスト分子の多様な選択が可能であり、設計の自由度が高いことか ら伝導度と安定性の高い新たな電解質として期待されている。その一方で、高い伝導 性を示すCOF の設計指針は未解明である。さらに、COF 空間内部におけるキャリア 分子の伝導機構や、COF 骨格が伝導機構に与える影響についても未だ明らかになって いない。機構解明には COF 骨格およびゲスト分子の原子レベルでのダイナミクスが 重要であるが、実験によって得られる情報は限られている。本研究では、量子化学シ ミュレーションを用いて伝導機構の解明を行った。
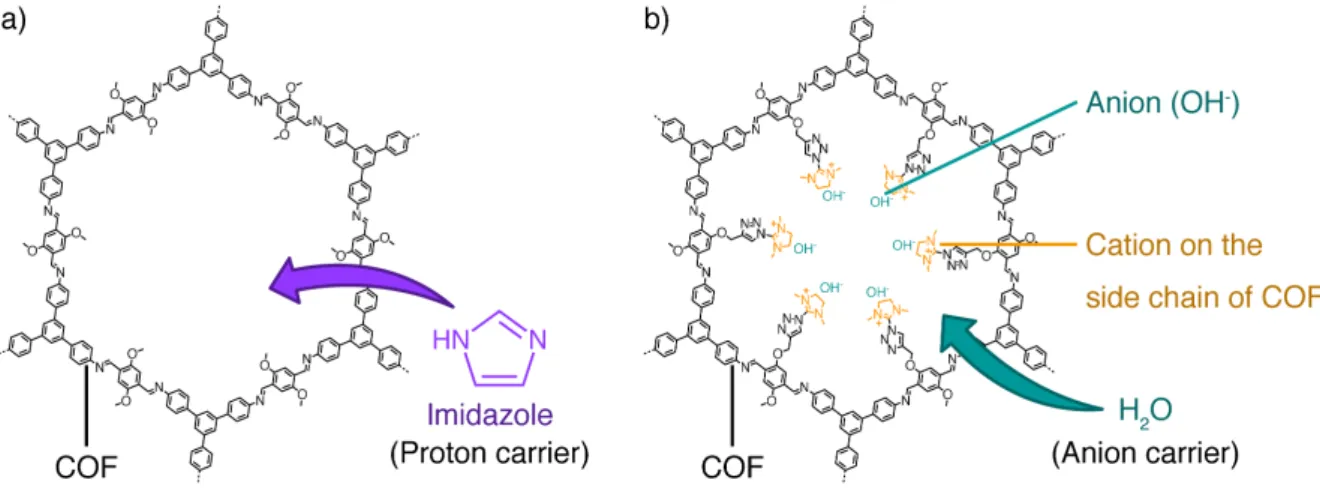
【計算方法】 キャリア分子と骨格の両方のダイナミクスを考慮した伝導機構の解明には、1000 原子以上を含む大規模系に対する十分に長時間のシミュレーションが必要である。そ こで、計算コストが低く高速計算が可能な量子化学的手法として、Density-Functional Tight-Binding (DFTB) 法と Divide-and-Conquer (DC) 法を組み合わせた DC-DFTB 法を 選択し、量子化学計算を用いたMolecular Dynamics (MD) シミュレーションを行った。 [1] 計算対象の系としては、イミダゾールをキャリア分子とするプロトン伝導性 COF (Fig. 1a) と、水をキャリア分子とするアニオン伝導性 COF (Fig. 1b) を選択した。[2] これらのCOF は 2 次元状構造の積層集積体であり、1 次元の細孔を有する。本研究で は周期境界条件のもとで、プロトン伝導性COF とアニオン伝導性 COF について実験 における測定温度である403 K と 353 K でそれぞれ DC-DFTB-MD シミュレーション を行った。プロトン伝導性COF では、プロトンを一つ追加した系での検討を行った。 【結果・考察】 COF 中におけるキャリア分子の分散に 基づく vehicle 機構と、キャリア分子間の プロトン移動に基づく Grotthuss 機構につ いて解析した。キャリア分子の平均二乗変 位 (MSD) の時間変化を Fig. 2 に示す。こ の傾きから得られた拡散係数はプロトン 伝導性COF では 1.39 × 10-5 cm2s-1、アニオ ン伝導性COF では 1.32 × 10-5 cm2s-1となっ た。後者の値は純粋な水の拡散係数6.59 × 10-5 cm2s-1より小さく[3]、COF の細孔内で 水分子の分散が制限されたことが示唆される。また、キャリア間のプロトン移動頻度 はプロトン伝導性COF では 95 ps 中に 2 回、アニオン伝導性 COF では 27 ps 中に 66 回と、系によって大きく異なることが確認された。 【参考文献】
[1] H. Nishizawa et al., J. Comput. Chem. 37, 1983 (2016). [2] H. Xu et al., Nat. Mater. 15, 722 (2016)
[3] A. J. Easteal et al., J. Chem. Soc., Faraday Trans. 1, 85, 1091 (1989)
Fig. 1. a) Scheme of the Proton-conductive COF. b) Scheme of the Anion-conductive COF.
京コンピュータを利用した非鉛化ペロブスカイト太陽電池の探索
1理研AICS○中嶋隆人,澤田啓介
Discovery of Pb-Free Perovskite Solar Cells via High-Throughput
Simulation on K Computer
○Takahito Nakajima, Keisuke Sawada RIKEN AICS, Japan
【Abstract】
In this study, we performed a systematic high-throughput simulation with density functional theory for 11,025 compositions of hybrid organic−inorganic halide compounds in ABX3 and A2BBʹX6 forms, where A is an organic or inorganic component, B/Bʹ is a metal atom, and X is a halogen atom. The computational results were compiled as a materials database. We performed massive computational simulation by using the K computer, which is a massively parallel many-core supercomputer in Japan. By applying the screening procedure to all the compounds in the materials database, we discovered novel candidates for environmentally friendly lead-free perovskite solar cells and propose 74 low-toxic halide single and double perovskites, most of which are newly proposed in this study. The proposed low-toxic halide double perovskites are classified under six families: group-14–group-14, group-13–group-15, group-11–group-11, group-9–group-13, group-11–group-13, and group-11–group-15 double perovskites.
【序】 ハイブリッド型有機–無機ハライドペロブスカイトは有望な次世代太陽電池材料の ひとつである.その変換効率は 2009 年の 3.8 %から急激にあがり,現在では 22 %を 超える.ハイブリッド型ハライドペロブスカイトの太陽電池としての顕著なパフォー マンスは,紫外可視領域での大きな吸収係数,高いキャリア移動,長い電子–正孔拡 散長,直接バンドギャップ遷移のような優れた光学特性と電気特性に由来する.太陽 電池材料として代表的なハイブリッド型ハライドペロブスカイトはメチルアンモニ ウム鉛ヨウ素 (MAPbI3, MA = CH3NH3) やホルムアミジン鉛ヨウ素 (FAPbI3, FA = HC(NH2)2) のような鉛化ハロゲン化合物である.これらの鉛化ペロブスカイトは低コ ストで容易に合成可能である.しかしながら,化学的不安定性や毒性の問題があり, 鉛を用いない非毒性元素からなるペロブスカイト材料の探索が望まれている.シミュ レーションは実験と比べて,ある化合物中の元素の置換を容易に実現することが可能 である.既に知られている化合物中の元素を別の元素に置き換えて,新しい機能を持 った材料を実験に先立って理論設計することができる.本研究では,京コンピュータ による元素戦略的な材料スクリーニングに基づいたマテリアルズ・インフォマティク ス手法により,非鉛化ペロブスカイト太陽電池の新規材料探索を実現した.とりわけ, 本研究の特徴は,京コンピュータを用いたハイスループットコンピューティングによ り,従来よりも多数の候補化合物に対しスクリーニングを実施した点にある.
【方法】
ABX3と A2BBʹX6型の化合物を考慮した.ここで,A = MA, FA, Cs; B/Bʹ = Be, B, C, N, Mg, Al, Si, P, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Sr, Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, In, Sn, Sb, Ba, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hg, Tl, Pb, Bi; X = Cl, Br, I であ る.全組み合わせ数は 11,025 である.平面波に基づいた密度汎関数法 (DFT) に基づ いた周期系計算により,下記のスクリーニング手続きにしたがって,非鉛化ペロブス カイト太陽電池の新規材料探索を実施した.計算には VASP プログラムを用いた. 1. PBE 汎関数を用い,構造最適化.ペロブスカイト構造を保持する化合物を採用. 2. PBE レベルで金属の系とバンドギャップが 3.5 eV 以上の化合物を除外. 3. スピン・軌道相互作用を考慮した HSE12 (SO-HSE12) 汎関数を使い,バンドギャ ップ計算.0.8 eV 以上かつ 2.2 eV 以下の直接遷移ギャップを持つ半導体を採用.間接 遷移ギャップを持つ半導体のうち,一番低い直接遷移ギャップとの差が小さい半導体 も採用. 4. 電子と正孔のキャリア伝導度が大きい半導体を採用. 5. 毒性元素 (Pb, Hg, Cd, As, Tl) を含む化合物を除外. 【結果・考察】 上記のスクリーニングにより,11,025 個の化合物から 74 個の低毒性非鉛化候補化 合物が残った.A2BBʹX6型に関しては,本研究により初めて提案された化合物である. B サイトに関して周期表の族の組合せで分類すると,下記の 6 タイプを提案すること ができる.詳細に関しては当日発表する.
(a) 14 族系化合物半導体: MA2GeSnI6 (1.56 eV) など (b) 13 族–15 族系化合物半導体: FA2InBiCl6 (1.37 eV) など (c) 11 族系化合物半導体: FA2CuAuBr6 (1.59 eV) など (d) 9 族–13 族系化合物半導体: FA2RhInI6 (1.63 eV) など (e) 11 族–13 族系化合物半導体: MA2CuInI6 (1.29 eV) など
古典動力学における時間階層性と非断熱過程:隷従原理と隷従者の反乱
京大福井センター 高塚和夫
Classical analog of quantum nonadiabatic transitions in molecular systems;
The slaving principle and uprising by the slaved.
Kazuo Takatsuka
Fukui Institute for Fundamental Chemistry, Kyoto University, Japan
【Abstract】Abstract in English (ca. 150 words).
Nonadiabatic dynamics arising from the breakdown of the Born-Oppenheimer approximation, which in principle separates the fast electronic motion from slow nuclear motion coexisting in a molecule, gives birth to various characteristic chemical reactions. We here study a classical analog of nonadiabatic dynamics. The notion of adiabatic elimination gives a basic principle frequently adopted in nonlinear physics, in which the mechanisms of generation of slow and large amplitude modes (or order parameters in statistical mechanics) are sought for. The slaving principle of Haken also rests on the adiabatic elimination methodology. It presents a scenario for slow modes to be evolved in time, eventually dominating the entire system and letting the many fast modes follow "faithfully". The slaving principle sets a theoretical foundation of many of natural and even social sciences. We here shed light on the characteristic roles of the fast modes that can result as a nonlinear recoil from the system regularity (order) formed by the slow modes and can trigger reorganization of the whole system dynamics, which may look like uprising by the slaved.
【序】分子の中では,原子核と電子の運動の圧倒的なタイムケールの違いによって, 量子論的な運動の階層性が生まれているとみなすことができる.この階層性は,断熱 性とその破れ(非断熱性)で特徴づけられる[1].同様に,タンパクや,内部に時間変 化するクラスター構造を持つ液体などには,速い運動モードと遅いモードを同時に内 包しているものがあり,特徴的な現象を示すことがある[2].このように,古典動力学 にも,動力学的階層性の形成とその破れが起きることがある.このような階層性動力 学の特徴的な現象として,安定に推移しているように見える遅い運動モードが,突然 大きな質的あるいは構造的変化を遂げる事が挙げられる.本講演では,時間的階層性 に関して,次の二点を議論したい.(1)古典系に於ける「断熱的動力学」と「非断 熱的遷移」を記述する運動方程式について.(2)その運動方程式から予想される現 象の一般論. 【理論】 統計力学,非線形力学などでは,時間スケールの異なる部分系からなる全 体集合において,遅いモードの成長と,それによって支配される全系のダイナミクス
の特徴の抽出の一般論やその応用研究が広く行われてきている.著名なものは,Haken による隷従原理で,何段階かによる力学的不安定化を経て,系の中に遅い不安定モー ドが成長し,安定で素早く対応することができる速いモードが,それに追随(隷従) することによって,系全体の特徴が遅い運動モードによって支配されていく(隷従原 理)というシナリオである.速いモードが断熱的に消去されていくプロセスは,一般 に「断熱消去」の方法によることが多く[3],Haken も隷従原理を断熱消去法によって 再定式化することを試みている[4]. 断熱消去法は,強力な一般論だが,遅いモードの成長とその結果の系全体での秩序 形成に主眼を置くため,速いモードが系全体に及ぼす影響を「遅いモードに働く平均 場」の形成程度に捉えがちである. 本講演では,古典力学の位相空間分布関数のための Liouville 方程式から出発して, 量子力学的非断熱遷移理論のアナログを「運動方程式」として取り出すことを試みる. その結果,速いモードが,一方的に遅いモードに追随するわけではなく,条件が整え ば,「反乱」を起こし,系全体の運動形態の転覆(大転換)の引き金を引くことが有 り得ることを示す. 古典系では,電子と原子核の運動のような量子力学的 entanglement が存在しないの で,馴染みの Born-Huang 展開はできない.そのため,速いモードと遅いモードを区 別するために,古典位相空間分布関数Γを,条件付き確率分布の形で表現する.
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
( ) ( )
, , , , , ; , , , , f f s s f f s s s s fs s t t t q p q p q p q p q p ここで,位置 q と運動量 p の方の添え字 f と s は速いモードと遅いモードを表し,
( ) ( )
, , s s s t q p は遅いモードの分布関数とする.ここから出発して得られる運動方程式 とその意味,簡単な応用例を講演で示す. 【参考文献】 [1] 分子の非断熱過遷移の理論は,通常,ポテンシャル面の乗り移り現象として,その遷移確率と便宜 的なホッピングモデルで議論されることが多い.我々は,電子波束動力学の立場から「非断熱電子動 力学理論」の提案と応用を行っている.“Fundamental approaches to nonadiabaticity: Towards a chemical theory beyond the Born-Oppenheimer paradigm.” T. Yonehara, K. Hanasaki, K. Takatsuka, Chemical Reviews, 112, 499 (2012).
“Chemical Theory beyond the Born-Oppenheimer Paradigm”, K. Takatsuka, T. Yonehara, K. Hanasaki, Y. Arasaki, (World Scientific, 2015).
また,本討論会(2017)での以下の講演を参考にしていただきたい:
2D03, 山本健太郎,高塚,“生物系から着想を得た単純な系における衝突誘起の逐次的な電子移動; 非 断熱の電子波束による研究”
3A05, 松岡貴英,高塚,“高強度レーザー中の非断熱電子波束の対称性の破れについて” [2] I. Ohmine and S. Saito, Acc. Chem. Res. 32, 741 (1999)
[3] C. W. Gardiner, Phys. Rev. A 29, 2814 (1984).
PubChemQCプロジェクト:
分子データベース構築と機械学習による電子構造の推定
1理研ACCC,2理研AICS
○中田真秀1,島崎智実2
PubChemQC Project:a large-scale first-principles calculation database and
estimation of electronic structure by machine learning
○Maho NAKATA1, Tomomi SHIMAZAKI2
1 Advanced Center for Computing and Communication, RIKEN, Wako, Japan 2 Advanced Institute for Computational Science, RIKEN, Kobe, Japan
【Abstract】Our aim is to create a database using quantum chemical calculations and create
new functional molecules from that data (http://ppcdb.org/). In recent years, with speech recognition, image recognition, etc., with the development of machine learning, highly accurate estimation close to humans has become possible. We wan to apply such approach to chemistry. In that case, we need large amounts of molecule data. Fortunately there are about 100 million molecules registered in PubChem (https://pubchem.ncbi.nlm.nih.gov/) and it is freely available. From the end of 2013 to May 2017, the structure optimization and the excited state energy by B3LYP/6-31G* are performed by TDDFT (B3LYP) /6-31+G* and accumulated about 4 million molecules of calculated data (Http://pubchemqc.riken.jp/). Using this huge amount of data, the electronic structure of the molecule was predicted by machine learning. For example, HOMO-LUMO gap had an accuracy of about 0.4 eV.
【序】 我々は量子化学計算を使いデータベースを作成し、そのデータから新規機能 分子の創出、提案を行うシステム構築を目指している(http://ppcdb.org/)。近年、音声 認識、画像認識などでは、機械学習の発達により人間に近い高精度な推定が可能にな ってきている。これを化学にも応用できないだろうか。その際には大量の分子のデー タが必要だが、幸いPubChem(https://pubchem.ncbi.nlm.nih.gov/)に 1 億分子ほど分子構 造が登録されており、自由に利用可能である。これに基づき我々は2013 年末から 2017 年 5 月に至るまで B3LYP/6-31G*による構造最適化および励起状態エネルギーは TDDFT(B3LYP)/6-31+G*で行い、400 万程度の分子の計算データを蓄積してきた (http://pubchemqc.riken.jp/)。この膨大なデータを用い機械学習により分子の電子構造の 予想を行った。HOMO-LUMO ギャップは 0.4eV 程度の精度であった。 【方法(分子の計算)】 PubChem(https://pubchem.ncbi.nlm.nih.gov/) に登録されている分子をすべてダウンロ ードし、CID (Compound Identification)、 分子の InChI 表記、分子の SMILES 表記、お よび分子量の表を作成した。分子量でソートし、小さい分子量をもった分子から計算 を始めた。分子の初期座標生成はInChI 表記の Open Babel を用いた。この初期座標を 用い、PM3、HF/STO-6G、B3LYP/6-31G* の順で構造最適化(量子化学計算)を行った。 計算パッケージにはSMASH, FireFly も用いつつ、最終の B3LYP/6-31G*レベルの計算 はGAMESS に依った。引き続き、この構造を用い、TDDFT の計算を行い、10 個の低 いエネルギー状態の励起状態を求めた。これらのうち正常に計算が終了したものを、 http://pubchemqc.riken.jp にアップロードしている。分子については、PubChem に登録 されている、純物質(混合物でない、SMILES 表記で”.”を含まない物質)、元素の種類 としては6-31G*でサポートされている H-Kr をそれぞれ計算対象とした。
【方法(データベース構築)】 まず得られた分子に対してInChI によるバリデーションを行った。構造最適化によっ て構造が変化し、分子としての解釈が変化することが有るためである。バリデーショ ンの可否については、PubChem に登録されている InChI と、構造最適化によって得ら れたInChI の化学式サブレイヤーおよび原子のコネクションレイヤーを比較して一致 した分子を可とした。計算結果から、軌道エネルギー、HOMO-LUMO ギャップ、励 起エネルギー、双極子モーメントなどを抽出し、PostgreSQL データベースで管理を行 った。 【方法(機械学習)】 今回はSMILES 表記の分子から、HOMO-LUMO ギャップなど電子構造を予想するこ とを行った。これにはSMILES 表記を RDKit に含まれる 1024bit を分子の特徴ベクト ルとした Toplogical fingerprint に変換し、それから scikit-learn ライブラリを用い、 support vector machine(SVM)および Gaussian radial basis function (RBF)をカーネルとし て機械学習を行った。教師データとしては InChI バリデーションを行った分子で、 4.5-6.5eV まで HOMO-LUMO ギャップが一様に分布するよう、ランダムに 2 万分子選 んだ。そこから98 万分子の HOMO-LUMO ギャップの予想を行った。 【結果・考察】 分子計算: 理研 RICC クラスタなどを用い、一日数千分子の計算が可能であった。ま た、InChI 表記には曖昧さがあり、PubChem データーベースには固体、混合物、ラセ ミ体なども登録されていたため、量子化学計算をこのような化学のデータベースに直 接厳密に適用するのは、できないし、また、意味もない。また、InChI 表記などの限 界により金属錯体なども必ずしも正しく表現できない場合があった(例としてはフェ ロセン)。これらをどう解決するかは今後の課題である。 データベース構築: 左図は、220 万分子について HOMO-LUMO ギャップを横軸に励起エネルギーを縦軸 にその相関をプロットしたものである。これから励起状 態は HOMO-LUMO ギャップとよい相関に有ることが解 った。尚、約 10%の分子がバリデーションに失敗した。 分子検索エンジンもhttp://pccdb.org/ で公開中である。 機械学習:下図はHOMO-LUMO ギャップを SVM および Ridge regression によって予測した結果である。RMSE は 0.3〜0.4eV 程度と、SMILES のみを用いて予測したが、 TDDFT による結果とよく一致した。詳細は当日発表する。
【参考文献】
[1] Maho Nakata and Tomomi Shimazaki, "PubChemQC Project: a Large-Scale First-Principles Electronic Structure Database for Data-driven Chemistry", J. Chem. Inf. Model., 2017, 57 (6), pp 1300-1308.
[2] Nakata Maho, "The PubChemQC project: A large chemical database from the first principle calculations", AIP Conf. Proc. 1702, 090058 (2016).
Photoactive yellow protein発色団の構造変化に対する高圧力の効果
京都大学 触媒・電池元素戦略ユニット ○福田良一
Isomerization of photoactive yellow protein chromophore
under high pressure
○Ryoichi Fukuda
Elements Strategy Initiative for Catalysts and Batteries, Kyoto University, Japan 【 Abstract 】 Pressure dependence of the energetics for the isomerization reaction of photoactive yellow protein chromophore is investigated with quantum chemical calculations using XP PCM (eXtreme Pressure Polarizable Continuum Model). The XP PCM considers the high pressure effect by the model repulsion potential between solute’s electrons and the PCM cavity surface. We consider the one-bond flip (OBF) and hula-twist (HTw) mechanisms for the isomerization. The OBF is energetically favorable, but it requires larger activation and reaction volumes than the HTw path. Consequently the OBF isomerization path would be suppressed under high pressure in the excited state.
【序】cis-trans 異性化は一段階で分子構造を大きく変えることができるため、光機能 性分子材料によく利用され、また、生物の光受容体においても cis-trans 異性化を伴う 応答メカニズムが見出されている。異性化反応による構造変化が大きいことは、反応 体積や活性化体積が大きい事を示唆する。cis-trans 異性化の反応速度に対する圧力効 果が報告されており、反応機構が圧力などの環境効果により変化する可能性がある。 Photoactive Yellow Protein (PYP)は細菌が青色光を感知するために働き、その発色団は p-クマル酸チオエステルである。基底状態(暗状態)で trans 位にあるヒドロキシフェ ニル基とカルボキシル基が、青色光を吸収すると cis 位に異性化する。タンパク中で は、体積変化の小さな経路を経て異性化している事が報告されており[1]、気相中や溶 液中における p-クマル酸エステルの異性化とは異なる経路を経ている可能性がある。 我々は、凝集相での高圧力効果を量子化学に取り込む方法である XP PCM (eXtreme Pressure Polarizable Continuum Model)を開発している[2]。本研究ではこの方法で、PYP 発色団の異性化に対する高圧力の効果を計算した。反応体積および活性化体積を求め、 反応経路の圧力依存性を考察し、異性化反応への環境効果に関する知見を得る。 【方法】XP PCM では、溶質分子を、各原子を中心とした球体の重なりで構成される キャビティ内に配置し連続誘電体で囲む。連続誘電体は溶質分子の電子密度と自己無 頓着となるように分極することで、溶質-溶媒間の静電相互作用を記述する。さらに、 キャビティ壁面と溶質分子の電子密度との間に、パウリ反発を模した強い反発ポテン シャルを置くことで、高圧下で支配的となる溶質-溶媒間の交換斥力を記述する。 我々がターゲットとするのは溶質-溶媒相互作用(Vint )を含む Schrödinger 方程式
0
int H V E (1) であり、相互作用は静電相互作用V e とパウリ反発項Vrからなる。 int e r V V V (2)静電相互作用は従来の PCM と同様に決定し、パウリ反発項は r V r r r (3) のように、溶質分子の電子密度 r と障壁階段関数 r からなる。圧力 は系の自由エネルギーG の体積微分から求められるが、XP PCM では系の 体積と PCM のキャビティ体積Vcが連動すると考える。 er c p G V G V (4) 系の全自由エネルギーは、XP PCM により求めた量子化学的エネルギー (Ger)にキャビテーションエネルギー(Gcav)を加えて求められる。 tot er cav G G G (5) PYP 発色団の計算には Cys69 を N-メチル イソプロピルアミンでモデル化 した p-クマル酸チオエステルアニオン(右上図)を用いた。基本的に ωB97XD 汎関 数 cc-pVDZ 基底関数による DFT/TDDFT 計算を行い、構造最適化では適宜 Tamm- Dancoff 近似を用いた。電子状態の妥当性は SAC/SAC-CI 法と比較して確認した。溶 媒にはクロロフォルム(弱極性)を用い、平衡溶媒和(equilibrium solvation)を仮定した。 【結果・考察】異性化のメカニズムとして、C7-C8 の結合
軸が回転する One-bond flip (OBF)と C7 を中心に C1-C7-C8 結合がねじれる(すなわち、C1-C7 と C7-C8 が同時回転す る)Hula-twist (HTw)が考えられ、それぞれ、活性化エネル ギーと活性化体積が異なると考えられる(右図)。 計算された反応エネルギーを Table 1 に示す。基底状態(S0) でパウリ反発を考慮しない計算(0 GPa)では、trans-cis エネ ルギー差(ΔGt-c)は 0.17 eV で trans 体が安定であった。OBF と HTw 経路の活性化エネルギー(ΔG‡ OBF, ΔG‡ HTw)はそれぞれ 1.39 eV,1.92 eV であり OBF 経路の方が低エネ ルギーであった。高圧力をかける と、trans-cis エネルギー差はわず かに小さくなり、OBF と HTw 経 路の活性化エネルギー差も小さ くなる。励起状態(S1)では、OBF により C1-C7-C8-C9 が 90°回転した構造が最安定となり、trans 体とのエネルギー差 (ΔGt-m)は 0.40 eV (0 GPa)であった。ただしこの構造は高圧力をかけると著しく不安定 化し、4.4 GPa では trans 体より 0.09 eV 不安定となる。つまり、OBF による光異性化 経路は、周辺環境との強い相互作用により抑制され、よりコンパクトな経路が優先す ると考えられる。 自由エネルギーの圧力依存性から反応体積ΔV と活性化体積 ΔV‡を見積もった(Table 1)。 HTw は OBF よりコンパクトな経路で反応が進む。また、S1の構造緩和により体積が 顕著に増大する(+16.7 Å3)のは、結合のねじれに加え、フェニル環内の結合長変化、 Cys69 とカルボキシルとの間の水素結合の強度に起因する。 【参考文献】
[1] Y. K. Jing et al. Nat. Chem. 5, 212 (2013); K. Pande et al. Science 352, 725 (2016).
[2] R. Cammi et al. Chem. Phys. 344, 135 (2008); R. Fukuda, M. Ehara, R. Cammi J. Chem. Theory Comput. 11, 2063, (2015); T. Yang, R. Fukuda, R. Cammi, M. Ehara J. Phys. Chem. A, 121 4363 (2017).
Table 1. Reaction energetics (eV), reaction and activation
volumes (Å3) of PYP chromophore in the ground (S 0) and
lowest excited (S1) state under high pressure (GPa).
S0 state S1 state p/GPa ΔGt-c ΔG‡OBF ΔG‡HTw ΔGt-m 0 0.17 1.39 1.92 −0.40 1.0 0.18 1.47 1.98 −0.28 4.4 0.16 1.72 2.06 +0.09 ΔVt-c ΔV‡OBF ΔV‡HTw ΔVt-m −0.4 8.0 4.8 16.7
モデルDNAのHartree-Fock計算
1城西大理,2九大院総合理工○寺前裕之1,青木百合子2
Hartree-Fock calculations of model DNA
○Hiroyuki Teramae1, Yuriko Aoki 2 1
Department of Chemistry, Faculty of Science, Josai University, Japan
2
Department of Molecular and Material Sciences, Interdisciplinary Graduate School of Engineering Sciences, Kyushu University, Japan
【Abstract】As an attempt at the electronic structure calculations of the B-type model-DNA, (poly-(guanine) poly-(cytosine)) double helix including sodium atoms as counter cation, hereafter referred as (poly-(dG)poly-(dC), double helix model polymer is treated by means of ab initio Hartree-Fock crystal orbital method adapting the screw axis-symmetry which results in great reduction of computational efforts. All sugar backbones and ions are included in the calculations. Energy band structures are calculated at 3-21G and 6-31G levels. It is significant that the bottom of the lowest conduction band at the Γ point of the first Brillouin zone has almost zero energy value. The effective masses of the hole and electron at the Γ point is 13.7 and 9.53, respectively. Both values are relatively large, therefore, the band conductions are expected not to be effective in this model-DNA backbone.
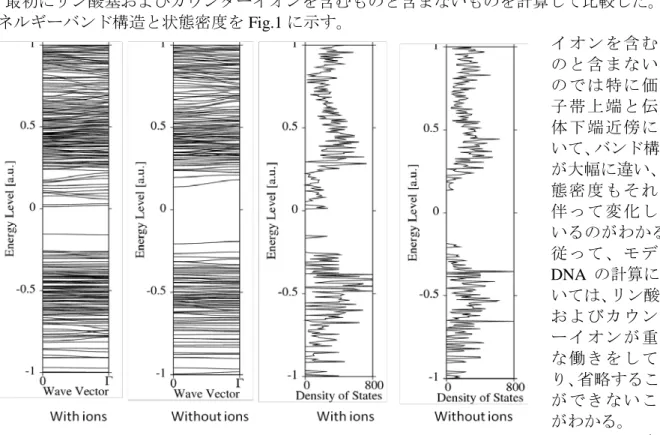
【序】 Ab initio 分子軌道法により、糖鎖とカウンターイオンまで含めた DNA の二重らせん 構造について、量子化学計算を行って生体高分子としてバルク状態での DNA の電子構造を求 めることは古くから検討されている。 DNA の計算を分子軌道法を基に行う場合には、tight-binding 法を使用したエネルギーバン ド計算による方法、いわゆる結晶軌道法がある。さらに DNA のようにらせん構造を有する一 次元高分子はらせん角を利用することで、エネルギーバンド計算が行えるが、このような大 規模計算は Otto, Clementi, Ladik による先駆的研究は存在するが、一本のらせん鎖またはリン 酸基およびカウンターイオンを含んでいないものに止まっていた[1]。 実際に計算を行っていくモデル DNA については、二重らせんモデルを対象としている。 DNA 二重らせんは塩基配列、塩基組成、相対湿度、カウンターイオンの組成や濃度により、 A, B または C 型などを取る。一般には水溶液中で安定化する B 型が多くの場合に見られるた め、本研究で考慮しているのも B 型である。 本研究ではこれまで有限クラスターとしてしか取り扱われていなかった DNA を無限系と して取り扱うことにより、その電子状態と導電性の関係などを解析することを目的とした。 【方法】 一次元結晶軌道法を用いた計算を行う。結晶軌道法では、Bloch 基底関数を用いる。 途中大幅に省略するが、実空間でのエネルギーの表現は次式で表される。 ここで H, F, P は実空間での一電子ハミルトニアン、Fock、電子密度を表し、通常の分子軌道 計算で用いられるものと同じである。ここでらせん対称性をポリマーが持っているとし、対 称軸が z 軸であるとすると、通常の p 軌道の代わりに pxおよび pyを次式のように、
とすることでより小さいユニットセルを用いて計算可能となる[2]。 基底関数は 6-31G を用い、最も簡単な DNA モデルとして、二重らせん状態のグアニン-シ トシンのペアのポリマーpoly-(dG)poly-(dC)を計算した。らせん角度は 36 度とした(10 ユニッ トで 1 ターン)[3]。 【結果・考察】 最初にリン酸基およびカウンターイオンを含むものと含まないものを計算して比較した。エ ネルギーバンド構造と状態密度を Fig.1 に示す。 イオンを含むも のと含まないも のでは特に価電 子帯上端と伝導 体下端近傍にお いて、バンド構造 が大幅に違い、状 態密度もそれに 伴って変化して いるのがわかる。 従って、モデル DNA の計算にお いては、リン酸基 およびカウンタ ーイオンが重要 な働きをしてお り、省略すること ができないこと がわかる。 表 1 には考慮 する隣接セル数 について全エネ ルギーなどの数値がどのよう に変化するかを示した。50 隣 接セルまで考慮してもエネル ギー値の収束はやや遅い。た だし、50 隣接セルを考慮する と伝導体下端の値が正から負 に転じており、伝導性を論じ る点では興味深い。 【謝辞】本研究は JSPS 科研費 16K05666 の助成によって行わ れた。 【参考文献】
[1] P. Otto, E. Clementi, J. Ladik, J. Chem. Phys. 78, 4547 (1983). [2] H. Teramae, Theoret. Chim. Acta 94, 311 (1996).
[3] Peng Xie, Hiroyuki Teramae, Kai Liu, and Yuriko Aoki, Int. J. Quantum Chem., 113, 489 (2013).
Fig.1 The energy band structures and the density of states of poly-(dG)poly-(dC) with
and without ions.
Table 1. Test calculations of poly-(dG)poly-(dC)
N total energy time[s] top of VB bot. of CB band gap 5 -3070.40145 10435 -0.15397 0.01266 0.16663 10 -3070.40179 20690 -0.15210 0.00906 0.16116 15 -3070.40243 30945 -0.15264 0.00722 0.15986 20 -3070.40277 40870 -0.15334 0.00539 0.15873 25 -3070.40314 51126 -0.15392 0.00396 0.15788 30 -3070.40347 61744 -0.15435 0.00267 0.15702 40 -3070.40419 81644 -0.15497 0.00050 0.15547 50 -3070.40502 102281 -0.15539 -0.00140 0.15399
フラグメント分子軌道法の多体展開とその物理学の描像
産総研・CD-FMatDmitri G. Fedorov
Many-body expansion in the fragment molecular orbital method and its
physical picture
○ Dmitri G. Fedorov
Research Center for Computational Design of Advanced Functional Materials (CD-FMat), National Institute of Advanced Industrial Science and Technology (AIST), Japan 【Abstract】
The physical nature of the terms in the many-body expansion in the fragment molecular orbital method is revealed in comparison to an isolated non-interacting reference state of fragments: polarization, charge transfer and exchange repulsion. Various size-extensive physical properties, such as atomic charges, fragment multipoles, energy, gradient, Hessian et cetera can be evaluated with the many-body expansion. The magnitude of many-body terms is demonstrated on various chemical systems, for instance, electron density is illustrated on a water trimer. 【序】 フラグメント分子軌道法(FMO)[1,2]では、巨大分子がフラグメントに分割され、フラ グメントとその多量体の量子化学計算から全系のエネルギー、解析勾配、解析二次微 分等は得られる。FMO 法では全系の物性のみならず、多体展開から得られる系の部 分的寄与も算出される。一体物性として、フラグメントの内部エネルギー、水和エネ ルギー、電荷を含む多極子モメント等がある。二体物性として、フラグメント間の相 互作用エネルギーや電荷移動等を挙げられる。 【理論】 示量的物性 A の FMO の M 体展開は
M m m M 2 1 A A A (1) 1 A は一体寄与である。
I I A A1 (2) M 体補正MA AM AM-1はフラグメントM 量体から計算される。
J I IJ A A 2 (3)
K J I IJK A A 3 (4) エネルギーA , M=2 の場合、 E
J I IJ I I E E EFMO2 (5) 全エネルギーは、各フラグメントの内部エネルギーとフラグメント間相互作用の和で ある。式1 は通常使われる FMO の多体展開である。相互作用しないフラグメントの 状態を参照状態(0)を導入すると、一体補正(参照状態との差)をも得られる。
M m m M 1 0 A A A (6) ここで分極項は
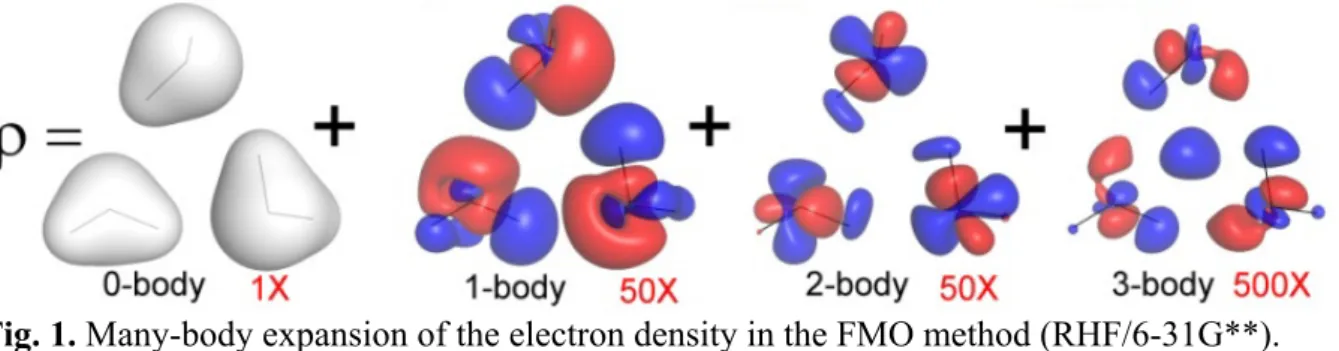
I 0 I I A A A 1 (7) しかし、FMO のフラグメント電子状態は、無撞着的に多体分極を取り込む為、一体 物性(1A)にも多体効果(分極)がある。多体効果MA(M>1)には、静電(分極)以 外の量子化学的多体効果が入る(即ち、電荷移動と交換反撥)。 【結果・考察】 具体的例として、水三量体の電子密度 を挙げる。
r
r
r
r
r
r 0 1 2 3 (8) 電子密度 は、孤立分子の電子密度
r 0
r 、分極による密度補正1
r 、電荷移動 と交換反撥二体(ペア)補正2
r と三体補正3
r からなる。Fig. 1. Many-body expansion of the electron density in the FMO method (RHF/6-31G**).
多局子モメント、二次微分、動力学の動径分布関数等の多体効果を議論する。 式1 の展開は M=N(N はフラグメントの個数)の場合、第一原理の値になるが、M<N で は、近似である。M 体展開の FMOM の誤差は、無視した M 体以上の量子効果に因む。 水集合体等、その効果が大きい系では FMO の誤差も大きくなる傾向があるが、誤差 相殺の影響もある。 【参考文献】
[1] D. G. Fedorov, WIREs, Comp. Mol. Sc., 印刷中。 [2] http://staff.aist.go.jp/d.g.fedorov/fmo/main.html。