医薬品医療機器総合機構信頼性保証部(〒1000013 東 京都千代田区霞が関 332 新霞が関ビル)
e-mail: ito-kanako@pmda.go.jp
本総説は,日本薬学会第 138 年会シンポジウム S14 で 発表した内容を中心に記述したものである.
2019 The Pharmaceutical Society of Japan
―Symposium Review―
GLP の制定の経緯,必要性及び基礎研究における信頼性に関する課題
伊藤かな子,染 谷 仁
Good Laboratory Practice: Initial Development, Necessity,
and Issues of Data Reliability in Basic Research
Kanako Itoand Hitoshi Someya
O‹ce of Non-clinical and Clinical Compliance, Pharmaceuticals and Medical Devices Agency (PMDA); Shin-Kasumigaseki Bldg., 332 Kasumigaseki, Chiyoda-ku, Tokyo 1000013, Japan.
(Received September 20, 2018)
This review describes the initial development of good laboratory practice (GLP) and follows the discoveries of quality control problems in labs that conducted tests in U.S. pharmaceutical companies. In addition to introducing the essence of the GLP standards, how the GLP ensures the reconstructability and reproducibility of study results is ex-plained in detail. Issues in nonclinical safety studies in drug development and approaches of the Japanese Pharmaceuti-cals and Medical Devices Agency to overcome them are also described. It is hoped that this review is helpful not only to those who work on drug development but also to faculties and students who work in academia and are involved in basic research when they attempt to resolve problems related to ensuring the reliability of basic research and research integrity. Key words―reconstructability; reproducibility; ensuring reliability; good laboratory practice; Pharmaceuticals and Medical Devices Agency
1. はじめに
独立行政法人医薬品医療機器総合機構(Phar-maceuticals and Medical Devices Agency; PMDA) は,医薬品,医療機器及び再生医療等製品の品質, 有効性及び安全性について,承認申請資料を審査 し,市販後における安全性に関する情報の収集,分 析,提供を通じて,国民保健の向上に貢献すること を 目 的 と し て , 平 成 16 年 4 月 に 設 立 さ れ た . PMDA信頼性保証部では,医薬品等の承認申請資 料に添付された資料が,厚生労働省の定める基準に 従って作成されたものであるかどうか,調査を行っ ている.
本 稿 で は , 米 国 に お い て good laboratory prac-tice(GLP)制定の契機となったサール社事件及び IBT 社事件を紹介するとともに,日本の製造販売承 認申請に関する厚生労働省の定める基準の 1 つであ る医薬品等の安全性に関する非臨床試験の実施の基 準に関する省令(GLP 省令)について,その概略 を紹介する.特に,GLP 省令が目的とする試験の 再構築と結果の再現性を可能とするための様々な仕 掛け(遵守事項)について解説する.さらに,医薬 品等の開発における非臨床安全性試験の問題点,そ の改善に向けたアプローチについて PMDA の取り 組みも合わせて紹介する.なお,本稿は筆者の個人 的な意見も含まれるため,すべてが PMDA の見解 ではない. 2. あなたの実験データは大丈夫 2-1. 実験データの品質管理 近年,臨床研究 データの改ざんや投稿論文のデータねつ造等,研究 不正に関する様々な問題がニュースとして一般紙に も取り上げられている.研究・実験データの改ざん や捏造を防止する方策はいくつか考えられるが,そ の中の 1 つに,データの「品質管理」という考え方 がある.すなわち,データに不正が発生しないよう にあらかじめ適切に作成工程を管理しながらデータ を取得するという考え方である.「データの品質管 理」の意味合いについて,「データ」と「品質管理」 はいずれも漠然とした用語のため,身近な例とし て,多くの方が利用するスマートフォンを取り上げ
て説明する.購入前のスマートフォンに対しては, 製造工場や携帯ショップで様々な品質管理を行って いる.その管理項目は,適切に製造されたか,設計 書通りの製品は作られたか,動作性は問題ないか, といった様々なチェック項目に合格して初めて出荷 され,使用可能になる.このスマートフォンを実験 データに置き換えて考えてみると,そのチェック項 目は違うものの,実験が計画通りに実施されたか, 正確なデータは得られたか,実験過程や実験結果が 適切に記録されたか,考察はされているか,仮説は 立証されたか,といった項目をクリアして初めて利 用可能な実験データが得られたことになる.本稿で は,このように,目的とするデータを得るためにす べての工程を適切に管理することを「データの品質 管理」と呼ぶこととする.また,この,「データの 品質管理」を実行するためには,品質を管理するた めのシステムを構築することが重要である.本稿に おける品質管理システムとは,データの品質を確保 するために,適切な体制を構築し,包括的なプロセ スの下で研究・実験を実施する一連のシステムを意 図している.データの品質管理システムには様々な ものが存在するが,以下に,実験室にて作成される データの品質管理を目的とした GLP について説明 する. 2-2. GLPのシステム GLPとは good labo-ratory practice の頭文字で,動物実験などのうち, 医薬品,医療機器,農薬,飼料添加物,動物用医薬 品,動物用医療機器,化学物質などの承認申請や登 録申請のために行われる非臨床安全性試験実施に関 する基準のことである.非臨床安全性試験データを 公的に通用させるため,対象となる被験物質の安全 性に関するデータの信頼性(再現性と客観性)の確 保を図ることを目的としている.つまり,安全性試 験を行う「試験施設」に適用される「優良な試験施 設の基準」である.具体的には,動物を用いた安全 性試験を計画・実験し,それを記録,最終報告書を 作成し,関連資料を保存する一連の流れの各段階で それぞれ,明確な目的や手順が計画されているか, 適正な実験工程で実施されているか,再構築・再現 可能なデータが得られているか,最終報告書は生 データが適切に反映されているか,試験関係資料が 適切に保存管理されているか等について,各段階に 対する監視体制が構築され,信頼性が確保されてい るこ と を達 成 する ため の 基準 と言 え る. なお , GLPは動物などを用いた試験に対する用語である が,医薬品等の開発におけるヒトの臨床試験には clinical の 頭 文 字 を あ て た good clinical practice (GCP),医薬品の製造には manufacturing をあて た good manufacturing practice(GMP)などがあ り,いずれも広い意味ではデータの信頼性の確保を 目的としたデータの品質管理システムである.
2-3. GLP制定の経緯 GLPは日本固有の制
度ではなく,その制定の発端は米国の医薬品等の審 査機関である医薬食品局(U.S. Food and Drug
Ad-ministration; FDA)に提出された製薬企業のデー タ不正であった.1972 年,FDA に提出された抗菌 剤の承認申請資料に多数の不備(サマリーに合わせ た生データの修正,恣意的なデータの抽出等)が認 められた(サール社事件).その後の 1975 年には, FDAに提出された抗炎症薬の承認申請資料に疑義 が生じたため,米国当局が査察を実施した結果,不 適切な行為(データねつ造,死亡動物の隠蔽,誤っ た被験物質の投与等)が認められた(IBT 事件). そこで,1978 年に米国は GLP を定め,1979 年よ り適用を開始した.動物を用いた安全性試験は,医 薬品や米国に限らず,世界各国において様々な工業 製品を対象として実施されていることから,米国が GLPを定めたすぐ後の 1981 年に,主要な先進国が 加盟する国際機関である経済協力開発機構(Or-ganisation for Economic Co-operation and Develop-ment; OECD)が GLP 原則を定めた.日本を含む OECD加盟各国間で GLP データの相互認証が確立 され,現在はこの OECD の GLP が国際的な基準 となっている.日本では,1982 年に当時の厚生省 が GLP 基準を文書化し,翌 1983 年に医薬品の製 造販売承認申請に対して求める基準として適用を開 始した. 2-4. GLP省令 以下に,日本の医薬品等の 製造販売承認申請に適用される GLP 省令の条項を 示す.当該省令は第 1 条から 19 条で構成され,動 物(微生物や細胞を含む)を用いた試験,特に医薬 品等の安全性を確認するための非臨床試験を実施す る施設に遵守が求められている. 総則(第 1 条第 4 条) 職員及び組織(第 5 条第 8 条) 試験施設及び機器(第 9 条第 10 条)
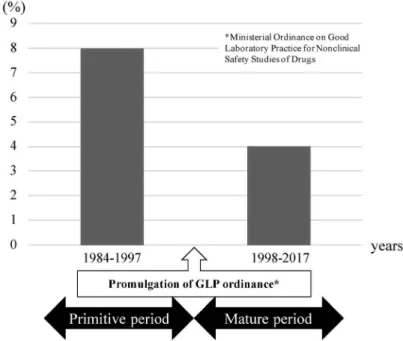
試験施設等における操作(第 11 条第 12 条) 被験物質等の取扱い(第 13 条第 14 条) 試験計画書及び試験の実施(第 15 条第 16 条) 報告及び保存(第 17 条第 18 条) 複数の場所にわたって実施される試験(第 19条) 上述の通り,GLP 省令は,医薬品等の製造販売 承認申請に係る非臨床安全性試験に適用される規制 要件であるが,各条に定められた遵守事項の中に は,研究・実験データの不正発生を防止するため に,基礎研究分野にも十分活用できると考えられる 事項がある.以下にその一部を示すが,これらは省 令の条文を平易な文面に変えているため,詳しくは 医薬品の安全性に関する非臨床試験の実施の基準に 関する省令(平成 9 年厚生省令第 21 号),医療機器 の安全性に関する非臨床試験の実施の基準に関する 省令(平成 17 年厚生労働省令第 37 号)又は再生医 療等製品の安全性に関する非臨床試験の実施の基準 に関する省令(平成 26 年厚生労働省令第 88 号)を 参考にされたい. データの記録(第 16 条) 生データの記録は,容易に消すことのできない 方法で,直接,直ちに,かつ,読みやすく記録 すること. コンピュータで記録する場合,入力日・入力者 等を記録すること. 生データを訂正する場合,訂正理由,訂正者, 訂正日を記載するとともに,訂正前のデータを 不明瞭にしない方法で訂正すること. 試験中の異常や予見できない事態が発生した場 合,速やかに責任者に報告し,改善措置を講じ て,経緯を記録すること. 資料保存施設(第 18 条) 資料保存施設(例えば,資料保存専用のキャビ ネット,部屋,建物,コンピュータの設置な ど)を設置し,そこで,生データ等を保存する こと. 資料保存施設では資料の確実な保存と検索がで きること. 資料保存施設の管理の責任者を設置し,その者 が許可した者以外は,資料保存施設に立ち入る ことができないこと. 資料保存施設からの資料の出し入れと移動は適 切に記録すること. 試験施設の体制(第 610 条) 試験施設の運営全般に責任を持つ「運営管理者」 を指定すること. 試験毎に各試験の実施と結果に責任を持つ「試 験責任者」を指名すること. 第三者的に実験操作やデータの監査を行う「信 頼性保証部門」を設置すること. 試験に使用する機器は,適切に保守点検,清 掃,修理を実施して,その内容を文書に記録し て保存すること. 2-5. GLP の導入効果 GLP制度の変遷 1978 年は米国が GLP の基準を発出した年である が,同年に日本でも GLP 制度の設立の動きが始 まった.日本では 1978 年に,まず,GLP 案の作成 を目的とした GLP 検討委員会が設置され,その後 の 1982 年には現在の GLP 省令の基礎となる「医 薬品の安全性試験の実施に関する基準」(昭和 57 年 3 月 31 日付薬発第 313 号厚生省薬務局長通知)が 発出された.この GLP 基準の発出により,GLP 試 験を実施する試験施設には GLP の各項目の適用を 求め られ る こと と なっ た. そ の基 準に 基 づい て 1984年より規制当局による GLP 査察が開始され, 1988 年に「医薬品 GLP 及び査察に関する規定の改 正について」(昭和 63 年 10 月 5 日付薬発第 870 号 厚生省薬務局長通知)が発出された.GLP 査察と は,各施設におけるこの GLP 適用状況を公的機関 (関係規制当局)によって確認することを指す. 1997 年には,より法的拘束力の強い「医薬品の安 全性に関する非臨床試験の実施に関する省令」(平 成 9 年厚生省令第 21 号)が公布された. GLP の導入効果 規制当局による GLP 査察では,査察の結果,規 定から逸脱が認められた場合,その内容や影響範囲 に応じて,改善を求め,GLP 適合又は不適合を判 断する.この査察による評価結果について,重大な 逸脱が認められた施設の割合について,GLP を導 入した初期である「創成期」(GLP 省令公布以前) とそれ以降の「成熟期」に分けて比較すると,明ら かな減少が認められ,現在では試験に信頼性を及ぼ すような重大な逸脱が認められる事例は極めて少な
Fig. 1. The Rate of Facilities with Signiˆcant Deviations くなっている(Fig. 1). 3. 信頼できるデータとは 上述の通り,医薬品等の製造販売承認申請に用い られる非臨床安全性データには,GLP という厳格 なデータ品質管理システムの下で作成されることが 求められているが,このような製造販売承認申請に 用いられるデータには,非臨床安全性試験だけでは なく,その他の試験(例えば,薬効薬理や非臨床薬 物動態など)データにも一定の品質管理が求められ ている.したがって,将来的な製造販売承認申請を 目指して実施される基礎研究においては,これらの 規制要件を認識することが,基礎研究から臨床応用 までの橋渡しを促進する上で重要である.以下に, 医薬品等開発の規制の概略を紹介するとともに,基 礎研究の段階から適切なデータを取得することの重 要性とその理由を説明する. 3-1. 医薬品等開発の落とし穴 医薬品等が実 用化されるためには,基礎研究から始まり,応用研 究,動物を用いた試験,治験,すなわちヒトでの臨 床試験を経て,厚生労働省の承認を取得するという 長い道のりがある.この道のりの中には,デスバ レー(死の谷)と呼ばれるいくつかの障壁(実用化 に至る前に脱落する原因)がある.その障壁には, 研究開発の資金不足や人材不足,知的財産戦略の欠 如,市場性を考えた開発戦略の欠如もあるが,規制 への理解不足が含まれる.前述の通り,製造販売承 認申請に用いられるデータには,GLP を含めた信 頼性に関する様々な基準に従ったものが必要とされ るものの,データを提出する段階になって,基準を 満たしていないことが発覚する場合などが該当す る.特に,医薬品等を開発する場合,様々なシーズ が含まれる基礎研究のデータを適切に取得すること により,製造販売承認申請のデータとして直接的に 活用できる可能性が高まる.つまり,基礎データを 取得する段階から信頼性が高く,品質管理された データを取得することが重要な鍵となるため,基礎 研究分野の方にもデータ取得後にどのようなステッ プが控えているかを理解したうえで,実験を実施す ることが有用と考える. 医薬品等の開発の各段階で収集したデータには, 信頼性に関する様々な基準への適合が求められる. 前述の通り,GLP 試験は,動物を用いた試験の中 でも特に安全性を確認するための試験,いわゆる非 臨床安全性試験に適用される.また,臨床試験は GCP,市販後調査は good post-marketing study practice(GPSP)が適用される.さらに,日本に おいては,安全性以外の非臨床試験(すなわち,薬 効薬 理 や薬 物 動態 試験 な ど) や品 質 試験 には , GLPや GCP は 適 用 さ れ な い も の の , い わ ゆ る 「信頼性の基準」(後述)と称される基準が適用され る.このように,日本では,医薬品等の製造販売承 認申請のために提出されるデータには,基本的にす
べてなんらかの信頼性に関する基準が適用される仕 組みになっている. 3-2. 信頼性の基準と GLP 上述の「信頼性 の基準」とは,医薬品,医療機器等の品質,有効性 及び安全性の確保等に関する法律施行規則(昭和 36年度厚生省令第 1 号)の第 43 条等に示された基 準であり,以下に示す 3 つの事項(データの正確 性,完全性・網羅性,保存性)が求められている. GLP省令では 19 条にわたり様々な事項の遵守を求 めているのに対し,信頼性の基準では信頼性を確保 するための必要最低限の事項が規定されていると言 える.つまり,前述した GLP による要求事項の一 部である「データの記録」,「資料保存施設」,「試験 実施の体制」といった GLP のエッセンスを基礎研 究等の実験でも採用することにより,製造販売承認 申請に求められる「信頼性の基準」を満たす可能性 が高まる. 〈信頼性の基準の概略〉 1)資料は,これを作成することを目的として行 われた調査又は試験において得られた結果に基 づき正確に作成されたものであること.(正確 性) 2)品質,有効性又は安全性を有することを疑わ せる調査結果,試験成績等が得られた場合に は,検討及び評価が行われ,その結果は当該資 料に記載されていること.(完全性・網羅性) 3)資料は,承認を与える(又は与えない)日ま で保存されていること.(保存性) 3-3. PMDA の業務 最後に,日本において 医薬品等に係る GLP や信頼性の基準等の調査(査 察)を実施している機関である PMDA を紹介する. PMDAは,以前,厚生労働省が行っていた業務の 一部を実施するために設立された独立行政法人であ る.したがって,GLP 査察も GLP 制度の創成期は 当 時 の 厚 生 省 が 行 っ て い た が , 現 在 は , 主 に PMDA が行っており,厚生労働省は必要に応じて 査察を実施することとなっている. PMDA では,GLP 調査のみならず,医薬品等の 製造販売承認申請前の段階の相談から,承認申請資 料の審査,各種の信頼性に関する調査,安全対策, 副作用・感染による健康被害の救済など,医薬品等 の開発段階から市場流通後まで一連の過程に関する 業務を行っている.また,前述の医薬品等開発にお けるデスバレー(死の谷)に対する取り組みとして, 相談制度を設けて,医薬品等の開発の初期段階か ら,大学や研究機関,ベンチャー企業を対象に積極 的に協力する体制を整えている. 4. おわりに 本稿の前半では,研究不正が大きなニュースにな る中で,データを品質管理するシステムの一例とし て,GLP を紹介した.大学や研究機関等で基礎研 究を実施する場合にも GLP のエッセンスの適用 は,高品質,かつ,再構築性・再現性の高い実験 データを取得するために有用と考えられる.後半で は,医薬品等の開発において,長い道のりとその障 壁について解説した.この中でも,信頼性の高い実 験データを取得し,製造販売承認申請に直接利用可 能な実験データを得ることはデスバレーの克服につ ながると考えられる.本稿が,医薬品等の開発を目 指す方に限らず,アカデミアの教職員,学生の皆様 に,基礎研究の信頼性確保に関する問題点を意識す る契機となり,さらに,その解決策・予防措置の策 定や公正な試験・研究推進の参考となれば幸甚であ る. 利益相反 開示すべき利益相反はない.