単純ベイズ確率モデルの導入による天然変性蛋白質領域予測の改良
6
0
0
全文
(2) Vol.2010-BIO-21 No.34 2010/6/19. 情報処理学会研究報告 IPSJ SIG Technical Report. い予測精度を実現することを試みた.. SEQ 1qu4 1f3t 1szr 1njj 2tod 2p3e 3btn 2j66 2plk 2plj 2oo0 2on3 1hkv 1d7k 2o0t 1ko0 1knw 1hkw 2nv9 2nva 1twi 3c5q 2qgh 7odc 1tuf 2yxx. 2. データセット 本研究では,PISCES サーバ8) を用いて,2010 年 3 月時点の Protein Data Bank(PDB)9) から,残基数が 50 以上,解像度 2.5 オングストローム以下,R 因子 0.25 以下,相互の配列 相同性が 20%以下の X 線結晶構造のみを含むデータセットを構築し,予測器の訓練,及び テストに用いた.また,天然変性蛋白質領域の定義には PDB ファイル中の REMARK465 行に記述された欠失残基 (missing residue) を用い,それ以外の ATOM 行に原子座標が存 2) 在する残基を安定な蛋白質領域とした. また,各残基について単体時の溶媒露出表面積. (Solvent Accessible Surface Area, SASA) を DSSP プログラム12) を用いて計算し,その. 100 150 200 . ISQLERFADKAAGIARGLRLNPQVSSSSFDLADPARPFSRLGEWDVPKVERVMDRINGFMIHNNCENKDFGLFDRMLGEIEERFGALIARVDWVSLGGGI RES A. ...|..|......|.|||||------||********.........|..|....|.|||||||..........|...|..|..||......|..||||.|| 2.90 A. ...|..|......|.|||||------.|**********.......|..|......|||||||..........|...|..|..||......|..||||..| 2.00 A. ...|..|......|.|||||------..*************....|.........||||||...........|...|..|..||......|.|||||..| 2.15 A. ...|..|...|..|.|||||------.|**********.......|..|....|.|||||||..........|...|..|..||......|..|||||.| 2.45 A. ...|.........|.|||||------||********...................|||||||..........|...|..|...|......|..|||||.| 2.00 A. ...|..||........||||||****************...|...............|||||.................|...|........|||.|||| 1.99 A. ......|......|.|||||------....********.......|.......|.||||||||.........|...|..|..||......|..|||.||- 2.05 A. ...|..||.||..|||||||||....**.........--...|||............||||||.........|..||..||.........|.||||||.| 1.65 A. ......|........|||||..*****---*****.........|..||......|.|||||.......|...|..|...|............|||||.| 2.14 A. ......|........|||||..*****---****..........|..||......|.|||||.......|...|..|...|.........|..|||||.| 1.70 A. ...|..|...|..|.|||||------||.|...........|..||.........|.|||||||.|||...||...|..|..||......|..||||||| 1.90 A. ...|..|...|..|.|||||------|.**********.......|.........|.|||||..........|...|..|..||.........|||||.| 3.00 A. ......||.......|||||||.|.|...............|..|..........|.||||||...||...||..||................||||||| 2.60 A. ......|......|.|||||------|..................|.........|.|||||..........|...|..|..||.........||||..| 2.10 A. ...|..||.|.....|||||||.|.|...............|..|......|...|.||||||...||...||..|...|.............||||||| 2.33 A. |..|..||.....|||||||||..|........|||....||||.............||||||||-......|..|...|............||||.|.| 2.20 A. |..|..||.....|.|||||||..|.........||....||||....|........||||||||-.........|...|............||||.|.| 2.10 A. ......|..|.....|||||||.|.|...............||.|....|..|..||||||||..|||...||..||..|.............||||||| 2.80 A. ...|..|........|||||||.|.........------..|...|..|....|.|.||||||||.|.....|..||..|..||.........||||||| 1.95 A. ...|..|......|.|||||||.|.........------..|...|..|....|.||||||||||.|....||..||..|..||.........||||||| 1.80 A. ...|.........|||||||||.|...........|.....|..|...|......|.||||...........||..|...|............|||||.| 2.00 A. ......||.......|||||||...|.......||.....|||....|.....||||||||||||||.....|..||...|...........|||||||| 2.40 A. ......||.......|||||||...|.......||.....|||....|.....||||||||||||||.....|..||...|...........|||||||| 2.30 A. ...|..|...|..|.|||||------.|***********......|.........|||||||..........|...|..|..||.........||||..| 1.60 A. ...|..|......|||||||||.|...........|.....|..|...|......|||||||..........||..||..|...........||||||.| 2.40 A. ...............|||||||.|...........|.....|...............||||||.........|...|..|..........||||||.|.. 1.70. id. 0.192 0.192 0.192 0.192 0.190 0.187 0.186 0.185 0.182 0.182 0.182 0.182 0.181 0.179 0.178 0.177 0.177 0.173 0.171 0.171 0.170 0.170 0.170 0.169 0.168 0.166. E. 6e-40 6e-40 1e-39 5e-40 8e-39 2e-72 1e-31 1e-44 2e-35 2e-35 4e-39 3e-39 6e-52 2e-37 1e-50 1e-69 1e-69 1e-50 5e-23 2e-23 3e-67 7e-67 2e-67 4e-37 6e-66 9e-69. * 天然変性領域 - ギャップ領域 | 蛋白質コア . 非蛋白質コア. SASA が Chothia によって求められた各残基種毎の最大溶媒露出面積13) との比が 0.05 以 図 1 構造アノテーションを加えたマルチプルシーケンスアラインメント Fig. 1 multiple sequence alignment with strucural anotations. 下の残基を蛋白質中心部に位置し,特に安定な構造を形成するコア残基として定義した.こ のセットは 5,121 の蛋白質鎖からなっており,48,921 残基の天然変性領域と 1,002,947 残 基の安定な領域を含んでいる.. (2). 3. 予 測 手 法. 得られたホモログのセット内での冗長性を除くため,ホモログ間で BLAST により 配列の相同性を計算し,sequence identity が 0.9 以上のペアが存在した場合,クエ. 本研究での予測は予測対象である蛋白質のホモログを既知構造データベース中から探索. リとの配列相同性が低いものを取り除く.. し,ホモログに存在する天然変性領域にアラインされる予測対象蛋白質中の残基は,より強. (3). い天然変性傾向を持つと推定することによって行われる.これは,蛋白質ホモログ間で天然. クエリ側のギャップ領域にアラインされたホモログ側の残基を切り捨て,クエリ側に ギャップの無いマルチプルシーケンスアラインメントを構築する.. 変性蛋白質領域が進化的に保存されていることに基づいている.天然変性蛋白質領域が進化. (4). 的に保存されていることは従前の研究で明らかになっており,また,その保存割合はホモロ. ホモログの立体構造から変性領域,コア領域を同定し,ホモログ中の各残基に構造情 報のアノテーションを加える.. グ間の sequence identity と相関があることが明らかになっている10) .. 3.2 従来の予測手法(重み付き平均) 従来の予測手法では i 番目の残基の天然変性傾向 Di は以下の式によって上述のマルチプ. 3.1 マルチプルシーケンスアラインメントの構築. ルシーケンスアラインメントからその sequence identity による天然変性傾向の重み付き平. 実際の予測に当たっては,まず以下の手順でクエリとなる予測対象の蛋白質に対してマル. 均として計算された7) .. ∑n. チプルシーケンスアラインメントを構築し,そこに構造情報を加えることで,図 1 のような. Di =. (1). j=1. αj I j. を利用し,ホモログを. (1) n ここで n はホモログの数であり,Ij は j 番目のホモログの sequence identity である.ま. PDB 中から探索する.その際,フォールドレベルで構造が一致するホモログのみを. た,αj は j 番目のホモログ中で,クエリの i 番目の残基にアラインされた残基が天然変性. 対象とするため,アラインメントの expectation value が 0.001 以上のもの,及びク. 領域にあれば 1,そうでなければ 0 である.. 構造情報アノテーション付のマルチプルシーケンスアラインメントを構築する. 配列データベースに対する繰り返し 2 回の PDB-BLAST. 11). エリに対してのカバー率が 0.6 以下の不十分なものは除く.. 2. c 2010 Information Processing Society of Japan.
(3) Vol.2010-BIO-21 No.34 2010/6/19. 情報処理学会研究報告 IPSJ SIG Technical Report. P (H1 , H2 , . . . , Hn |dis) = P (H1 |dis)P (H2 |dis) · · · P (Hn |dis)P (dis). 3.3 単純ベイズ確率モデルによる予測 単純ベイズ確率モデルは,各特徴の条件付き独立性仮定と共にベイズの定理を適用する単. (4). また,同様にエビデンスについても,. P (H1 , H2 , . . . , Hn ) = P (H1 )P (H2 ) · · · P (Hn ). 純な確率的モデルである. このモデルによる判別は,条件付独立が成立せず,クラスの出現 する確率の推定に誤差がある場合でも,判別されるクラスまで誤る確率は低いことが示され. となる.. ており,単純なモデルでありながら広範囲の問題に対して良い予測性能を示すことが知られ. 結果,天然変性確率は. ている14) .本研究ではこの単純ベイズ確率モデルを用いて,構造情報のアノテーションが. P (dis|H) = P (dis). 付加されたマルチプルシーケンスアラインメントが与えられた際の各残基の天然変性確率. n ∏ P (Hj |dis) j=1. を求めることで,天然変性蛋白質領域の予測を行う.. (5). (6). P (Hj ). ここであるクエリ蛋白質について,そのホモログに対する構造情報アノテーション付きマル. となり,ある残基が天然変性領域に存在する場合に同じ特徴を持つホモログが出現する確率. チプルシーケンスアラインメント H が与えられた時,クエリ蛋白質の i 番目の残基が天然. と,その特徴を持つホモログの出現確率から計算可能となる.. 変性領域にある確率を考える.この場合の天然変性確率は条件付き確率 P (dis|H) である. この時,H に含まれるアラインメント全域からの情報を全て利用すると,必要となるホモ. が,この条件付き確率はホモログの数が不定であり,その数が大きい場合にクラスの数が非. ログの特徴クラスの分類が膨大になり,各特徴クラスでの頻度を計算することが困難とな. 常に多くなるため,データベースから計算することが困難である.. る.そこで,本研究では残基レベルの構造情報はホモログ中でクエリの i 番目の残基にアラ. そのため,まずこの確率をベイズの定理を用いて変形し,個々のホモログを別々に書き下す.. インされる残基のもののみを利用し,特徴クラスの分類を行った.本研究で用いたホモログ. P (H|dis)P (dis) P (H) P (H1 , H2 , . . . , Hn |dis)P (dis) = P (H1 , H2 , . . . , Hn ). の特徴は以下のものである.. P (dis|H) =. (1). クエリとの間のアラインメントの sequence identity. • Sequecen identity は 0.1 刻みで別々のビンとし,10 のクラスに分類. (2) (2). クエリの i 番目の残基にアラインされた残基の状態. ここで P (dis) は天然変性領域に残基が存在する事前確率であり,P (H|dis) は尤度,P (H). • 天然変性蛋白質領域. はエビデンスである.また,P (H) は各ホモログの特徴クラスであり,n はホモログの総数. • 蛋白質コア安定領域. である.. • 非蛋白質コア安定領域. ここで尤度を条件付き確率に書き換えて分解すると,以下の様になる.. • ギャップ領域 (ホモログ側にアラインされる残基が存在しない) ここでギャップ領域を独立したクラスとして扱っているが,これは Radivojac らの研究に より,天然変性蛋白質領域では配列の進化速度が他の安定な領域に比べ速く,より置換や欠. P (H1 , H2 , . . . , Hn |dis) = P (H1 |dis)P (H2 , . . . , Hn |H1 , dis). 失,挿入が起きやすいことが知られており15) ,ギャップ領域にアラインされた残基は安定領. = P (H1 |dis)P (H2 |H1 , dis)P (H3 , . . . , Hn |H1 , H2 , dis) .. .. 域にアラインされた残基に比べ,より強い天然変性傾向を持つと考えられるためである.最. (3). 終的に,全てのホモログの特徴は 40(= 10 × 4) のクラスに分類され, そして全てのクラス. この定式化では他の別のホモログの情報が与えられた場合の,あるホモログの出現確率を求. について,その出現確率と,ある残基が天然変性領域に存在する場合に出現する確率を訓練. める必要が有り,困難である.そこで,ここに条件付き独立性仮定を導入し,各ホモログの. セットから計算しておくことで,式 (6) によりホモログの構造情報アノテーション付マルチ. 特徴 Hj が他のホモログの特徴に依存せず独立である仮定とする.するとホモログの出現確. プルシーケンスアラインメントが与えられた時,各残基の天然変性確率を求めることが可能. 率は対象残基の状態のみに依存するようになり,. となる.. 3. c 2010 Information Processing Society of Japan.
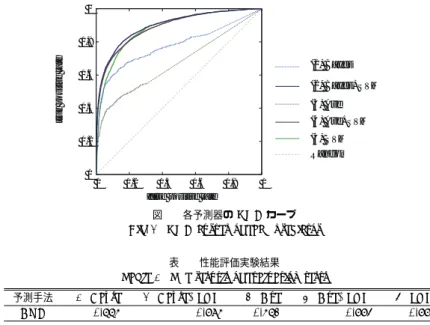
(4) Vol.2010-BIO-21 No.34 2010/6/19. 情報処理学会研究報告 IPSJ SIG Technical Report. 3.3.1 事前確率の推定. まま用いている.. 天然変性確率を求めるのに必要となる尤度とエビデンスについては,条件付き独立性仮定. また,ホモログの配列アラインメントを利用した予測手法の予測精度は既知構造データベー. を導入することで,データベースから容易に計算可能となることを示した.事前確率 P (dis). ス中にどれだけ配列相同性が高いホモログが存在するかに依存する.この性能評価ではより. も同様にデータベースから天然変性蛋白質領域の存在確率として求めることが可能である. 正確に性能を見積もるため,クエリに対してが sequence identity が 0.9 を超えるような非. が,その場合全ての蛋白質残基について事前確率の値は一定となる.この事前確率は残基種. 常に近縁のホモログについては予測の際に常に除外し,また,通常,予測を実行したい場合. 毎にその天然変性傾向に応じて異なる値を用いるなどの改良が可能であるが,本研究ではこ. には,あまり高い配列相同性を有する既知構造蛋白質が存在することは稀であることを考慮. の事前確率を局所的な配列特徴から機械学習を用いることでさらに高精度に推定すること. し,sequence identity が 0.5 以上のホモログを全て除外した場合でもテストを行った.. を試みた.事前確率の推定には,既に従前の研究で構築された教師付学習アルゴリズムで. 4.1 評 価 指 標. あるサポートベクターマシン (Support Vector Machine, SVM) に基づいた予測システム7). 天然変性蛋白質領域の予測は 2 クラスの分類問題であるため,通常は予測精度の評価指. を用いた.これは PSI-BLAST によってクエリ配列から変換された位置特異行列 (Position. 標にテストセット中で予測が正解となった割合である Q2 予測精度が用いられる.しかし,. Specific Score Matrix, PSSM) の予測対象となる残基の周囲 27 残基分を入力として学習,. 天然変性蛋白質領域の予測ではデータセット中に天然変性蛋白質領域が 5%程度しか存在. 予測を行うものであり,0 から 1 の間に正規化された天然変性傾向が予測値として返され. しないため,Q2 予測精度を評価指標に用いた場合,全ての残基を非天然変性領域として. る.しかし,予測システムの返す値はあくまで天然変性傾向であり,天然変性確率ではな. 予測することで容易に高い予測精度が実現できてしまう.そのため,本研究では予測の評. い.そのため,Ward らの方法. 16). を用い,あらかじめ訓練セットと非冗長のテストセット. 価指標として,Q2 予測精度ではなく,各擬陽性率における感度をプロットしたものである. から予測システムの返す天然変性傾向毎に実際の天然変性領域の存在確率を計算しておき,. ROC(Receiver Operator Characteristic) カーブ17) の下の面積である AUC(Area Under. それを利用して SVM の予測結果を確率に変換し,事前確率とした.. the Curve) を用いている.この指標は0から1の間の値をとり,ランダムな予測の場合に 0.5 となり,値が 1 に近づくほど良い予測結果であることを示す.. 4. 性 能 評 価. 4.2 実 験 結 果. 本研究によって提案された新たな手法の予測性能を評価するため,5 フォールドの交差検. 図 2 は交差検定法により得られた予測結果を用いて描かれた ROC カーブである.新た. 定法を用いて性能評価実験を行った.比較のため同じ訓練セットを用いて従来の予測手法. に提案された単純ベイズ確率モデルに基づいた手法による予測結果は全ての擬陽性率にお. (重み付き平均)と SVM に基づく機械学習による予測器を再構築し,以下の 5 つの手法に. いて従来の手法の結果より良い予測結果を返し,AUC では 0.1 近い改善がみられた.また,. ついてその性能を評価した.. 単純ベイズ確率モデルに機械学習により推定された事前確率を用いた場合,機械学習による. (1). 単純ベイズ確率モデル+一定の事前確率 (Bayes). 予測単体に比べて特に擬陽性率の低い領域について予測精度の改善が見られ,また,AUC. (2). 単純ベイズ確率モデル+機械学習により推定された事前確率 (Bayse+SVM). でも改善が見られた.従来の重み付き平均と機械学習による予測を組み合わせた手法でも機. (3). 重み付き平均 (Ave). 械学習による予測単体に比べ AUC の改善が見られているが,その改善の度合いは単純ベイ. (4). 重み付き平均+機械学習による予測 (Ave+SVM). ズ確率モデルに機械学習により推定された事前確率を用いた場合の方が優っており,今回提. (5). 機械学習による予測 (SVM). 案した単純ベイズ確率モデル手法がより優れた手法であることが示された.. 4 番目の手法は従前の研究で既に提案された手法で,ホモログの配列アラインメントから得. 4.2.1 高い配列相同性を有する既知構造蛋白質を除いた場合. られた予測の結果と機械学習による予測結果を重み付で足し合わせることで組み合わせた. 表 2 は配列アラインメントからの予測を行う際に sequence identity が 0.5 を超えるホモ. 10). 予測法であり,個々の予測結果より,良い結果が得られることが示されている. .ここで,. ログを全て除外し, より厳しい条件下で性能評価を行った場合の結果である.ホモログの構. 組み合わせの際の重みは今回の訓練セットに最適化せず,従来の研究で用いられた値をその. 造情報を利用した手法は,全てのホモログを利用した場合に比べその利用手法に関わらず,. 4. c 2010 Information Processing Society of Japan.
(5) Vol.2010-BIO-21 No.34 2010/6/19. 情報処理学会研究報告 IPSJ SIG Technical Report 1. なケースにおいても有用であることが示された.. 参. true positive rate. 0.8. (1) Bayes (2) Bayes+SVM (3) Ave (4) Ave+SVM (5) SVM. 0.2. Random 0 0. 0.2. 0.4 0.6 0.8 false positive rate. 1. 図 2 各予測器の ROC カーブ Fig. 2 ROC curves of each predictor 表 1 性能評価実験結果 Table 1 The resutls of evaluation tests 予測手法. AUC. (1) Bayse 0.776. (2) Bayse+SVM 0.896. (3) Ave 0.673. (4) Ave+SVM 0.885. (5) SVM 0.881. 明らかに性能の低下が見れらた.しかし,性能の低下は今回提案した単純ベイズ確率モデル を利用した手法においてより少なくなっており,また,機械学習による予測と組み合わせた 場合,まだ有意にその性能を向上させており,厳しい条件下であっても十分に有用であるこ とが示された. 表 2 性能評価実験結果 (sequence identity ≥ 0.5 のホモログ無し) Table 2 The resutls of evaluation tests (without homologs with sequence identity ≥ 0.5) 予測手法. AUC. (1) Bayse 0.716. (2) Bayse+SVM 0.889. (3) Ave 0.607. (4) Ave+SVM 0.883. 文. 献. 1) Tompa P.: Intrinsically unstructured proteins., Trends Biochem. Sci., Vol.27, pp. 523–533 (2002). 2) Dunker A.K., Brown C.J., Lawson J.D., Iakoucheva L.M., Obradovic Z.: Intrinsic dis and protein function., Biochemistry, Vol.41, pp.6573–6582 (2002). 3) Dyson H.J. and Wright P.E.: Intrinsically unstructured proteins and their functions., Nat. Rev. Mol. Cell Biol. , Vol.6, pp.197–208 (2005). 4) Prilusky J., Felder C.E., Zeev-Ben-Mordehai T., Rydberg E.H., Man O., Beckmann J.S., Silman I., Sussman J.L..: FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics, Vol.21, pp.3435– 3438 (2005). 5) Dosztanyi Z., Csizmok V., Tompa P., Simon I.: The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol., Vol.347, pp.827–839 (2005). 6) He B., Wang K., Liu Y., Xue B., Uversky V.N., Dunker A.K..: Predicting intrinsic dis in proteins: an overview., Cell Res., Vol.19, pp.929–949 (2009). 7) Ishida T. and Kinoshita K.: PrDOS: prediction of dised protein regions from amino acid sequence., Nucleic Acids Res., Vol.35, pp.460–464 (2007). 8) Wang G., Dunbrack R.L. : PISCES: a protein sequence culling server., Bioinformatics, Vol.19, pp.1589–1591 (2003). 9) Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I.N., Bourne P.E. : The Protein Data Bank., Nucleic Acids Res., Vol.28, pp.235–242 (2000). 10) 石田貴士, 中村周吾, 清水 謙多郎: アミノ酸配列によるタンパク質 dis 領域の予測, 物 性研究, Vol.86, pp.56–60 (2006). 11) Li W., Jaroszewski L., Godzik A.: Sequence clustering strategies improve remote homology recognitions while reducing search times. Protein Eng., Vol.15, pp.643– 649 (2002). 12) Kabsch W. and Sander C.: Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, Vol.22, pp. 2577–2637 (1983). 13) Chothia C.: The nature of the accessible and buried surfaces in proteins. J. Mol. Biol., Vol.105, pp.1–14 (1976). 14) Pedro D. and Pazzani M.: On the optimality of the simple Bayesian classifier under zero-one loss Machine Learning, Vol.29, pp.103–137 (1997). 15) Improving sequence alignments for intrinsically disordered proteins. Radivojac P.,. 0.6. 0.4. 考. (5) SVM 0.881. 5. ま と め 本研究ではアミノ酸配列からの天然変性蛋白質領域の予測精度を,ホモログの構造情報を 利用する際に単純ベイズ確率モデルを用いることで向上させた.その予測精度の向上は,配 列相同性の高い近縁のホモログが利用可能な場合において顕著であったが,あまり配列相同 性の高いホモログが利用できない場合においても十分な予測精度の向上が確認され,実用的. 5. c 2010 Information Processing Society of Japan.
(6) Vol.2010-BIO-21 No.34 2010/6/19. 情報処理学会研究報告 IPSJ SIG Technical Report. Obradovic Z., Brown C.J., Dunker A.K.: Pac Symp Biocomput., pp.589–600 (2002). 16) Ward J.J., Sodhi J.S., McGuffin L.J., Buxton B.F. and Jones D.T.: Prediction and functional analysis of native disorder in proteins from the three kingdoms of life J. Mol. Biol., Vol.337, pp.635–645 (2004). 17) Zweig M. H. and CampbellG.: Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin. Chem., Vol.39, pp.561–577 (1993).. 6. c 2010 Information Processing Society of Japan.
(7)
図
関連したドキュメント
It is suggested by our method that most of the quadratic algebras for all St¨ ackel equivalence classes of 3D second order quantum superintegrable systems on conformally flat
We present a Sobolev gradient type preconditioning for iterative methods used in solving second order semilinear elliptic systems; the n-tuple of independent Laplacians acts as
We use these to show that a segmentation approach to the EIT inverse problem has a unique solution in a suitable space using a fixed point
This paper develops a recursion formula for the conditional moments of the area under the absolute value of Brownian bridge given the local time at 0.. The method of power series
Answering a question of de la Harpe and Bridson in the Kourovka Notebook, we build the explicit embeddings of the additive group of rational numbers Q in a finitely generated group
The main problem upon which most of the geometric topology is based is that of classifying and comparing the various supplementary structures that can be imposed on a
“Breuil-M´ezard conjecture and modularity lifting for potentially semistable deformations after
Then it follows immediately from a suitable version of “Hensel’s Lemma” [cf., e.g., the argument of [4], Lemma 2.1] that S may be obtained, as the notation suggests, as the m A
