CTD 第 2 部
2.7 臨床概要
2.7.1 生物薬剤学試験及び関連する分析法
目次
頁 表一覧... 2 図一覧... 3 付録一覧... 4 略号及び用語の定義... 5 2.7.1.1 背景及び概観... 6 2.7.1.1.1 製剤開発の概観... 8 2.7.1.1.1.1 開発初期の製剤... 10 2.7.1.1.1.2 開発後期の製剤... 10 2.7.1.1.1.3 バニプレビルの溶解性及びバニプレビル製剤の崩壊及び溶出試験の 概略... 11 2.7.1.1.2 バニプレビル製剤のin vivo での挙動に関するまとめ ... 13 2.7.1.1.3 ヒト生体試料中の薬物濃度測定法... 14 2.7.1.2 個々の試験結果の要約... 15 2.7.1.2.1 バイオアベイラビリティ比較試験... 15 2.7.1.2.1.1 非日本人健康成人男女を対象としたバイオアベイラビリティ比較試 験(FFP 製剤 vs. 固形製剤)(006試験)... 16 2.7.1.2.1.2 非日本人健康成人男女を対象としたバイオアベイラビリティ比較試 験(FFP 製剤 vs. 新規 LFC 製剤)(014試験) ... 16 2.7.1.2.1.3 日本人健康成人男性を対象としたバイオアベイラビリティ比較試験 (FFP 製剤 vs. pPMF 製剤)(021試験)... 17 2.7.1.2.1.4 非日本人健康成人男女を対象としたバイオアベイラビリティ比較試 験〔PMF( )製剤vs. 新規 LFC 製剤〕(032試験) ... 17 2.7.1.2.2 バイオアベイラビリティ及び食事の影響に関する試験... 18 2.7.1.2.2.1 バニプレビルの絶対バイオアベイラビリティ評価試験(015試験) ... 18 2.7.1.2.2.2 日本人健康成人男女を対象とした食事の影響試験(049試験) ... 19 2.7.1.3 全試験を通しての結果の比較と解析... 20 2.7.1.3.1 バイオアベイラビリティ... 20 2.7.1.3.2 食事の影響... 22 2.7.1.4 付録... 25表一覧
頁 表2.7.1: 1 バニプレビル臨床開発における主要な製剤... 9 表2.7.1: 2 バニプレビルの臨床試験で使用した主要製剤の組成... 9 表2.7.1: 3 溶出試験方法と規格... 11 表2.7.1: 4 臨床試験使用製剤の主要バッチにおける崩壊時間... 12 表2.7.1: 5 バニプレビルフリー体の溶解性... 13 表2.7.1: 6 ヒト生体試料中のバニプレビル濃度測定法... 15 表2.7.1: 7 バニプレビル非標識体(経口投与)及び重水素標識体(静脈内投与)を併用し た際の血漿中薬物動態パラメータの要約統計量(015試験)... 19 表2.7.1: 8 日本人健康成人被験者にバニプレビル300 mg を PMF( )製剤で空腹時あ るいは食後単回経口投与した際の平均血漿中薬物動態パラメータの要約統計量 (049試験)... 20 表2.7.1: 9 非日本人(014試験)及び日本人(021試験)健康成人での pPMF 製剤と FFP 製 剤の血漿中曝露比較... 21 表2.7.1: 10 バニプレビルの薬物動態への製剤の影響に関する統合薬物動態解析結果... 22 表2.7.1: 11 バニプレビルの薬物動態に対する食事の影響... 23 表2.7.1: 12 バニプレビルの薬物動態への食事の影響に関する統合薬物動態解析結果... 24図一覧
頁 図2.7.1: 1 臨床試験に用いた製剤の概略と各製剤間の関連性... 7 図2.7.1: 2 バニプレビル(MK-7009、 )の化学構造... 8
付録一覧
頁 付録2.7.1: 1 バニプレビルの臨床試験で使用した製剤... 25 付録2.7.1: 2 生物薬剤学的試験の要約... 27 付録2.7.1: 3 生物薬剤学的試験の薬物動態データの表... 32 付録2.7.1: 4 バニプレビルの AUC0-∞に及ぼす食事の影響の要約... 41略号及び用語の定義
略語 定義
バニプレビル Vaniprevir 開発番号:MK-7009、
AUC Area under the drug concentration-time curve 薬物濃度-時間曲線下面積 BCS Biopharmaceutics Classification System 生物薬剤学分類システム
BHA Butylated Hydroxyanisole ブチルヒドロキシアニソール
BHT Butylated Hydroxytoluene ジブチルヒドロキシトルエン
b.i.d. Twice daily 1日2回
C12hr Plasma concentration at 12 hr postdose 投与後12時間の血漿中濃度
Caco-2 Human colon adenocarcinoma cell ヒト大腸癌細胞株
CI Confidence interval 信頼区間
CLp Plasma clearance 血漿クリアランス
Cmax Maximum drug concentration 最高薬物濃度
FCT Film coated tablet フィルムコーティング錠
FDA Food and Drug Administration 米国食品医薬品局
FFP 製剤 Fit-for-purpose formulation 開発初期製剤
FMI 製剤 Final market image 最終製剤
GMR Geometric mean ratio 幾何平均比
HCV Hepatitis C virus C 型肝炎
Glycerol ester of fatty acid グリセリン脂肪酸エステル
IV Intravenous administration 静脈内投与
LC-MS/MS Liquid chromatography-tandem mass spectrometry
液体クロマトグラフィー-タンデムマススペク トロメトリー法
LFC 製剤 Liquid filled capsule 液体充てんカプセル剤
nLFC 製剤 New liquid filled capsule 新規液体充てんカプセル剤
NS3/4 Non-structural 3/4A 非構造領域3/4A
PK Pharmacokinetic(s) 薬物動態
PMF 製剤 Preliminary market formulation 市販候補製剤
PMF( )
製剤
PMF used vaniprevir as drug substance バニプレビル を原薬とした市販 候補製剤 PMF( ) 製剤 PMF used vaniprevir as substance drug バニプレビル を原薬とした市販 候補製剤
pPMF 製剤 Prototype preliminary market formulation プロトタイプの市販候補製剤
P.O. Oral administration 経口投与
Polyoxyl 35 ポリオキシル35ヒマシ油
QC Quality check 品質確認
q.d. Once daily 1日1回
SEDDs Self-emulsifying drug delivery system 自己乳化させて薬物を送達するシステム
SD Single dose 単回投与
SOP Standard of procedure 標準操作手順書
Tween 80 Polysorbate 80 ポリソルベート80
t1/2 Elimination half-life 消失半減期
Tmax Time to the maximum drug concentration 最高薬物濃度到達時間
2.7.1.1 背景及び概観 バニプレビル(Vaniprevir、MK-7009、 )はHCV の複製に必須な酵素である NS3/4A プロテアーゼの強力な阻害剤である。バニプレビルはHCV 遺伝子型1の感染に対し、リバビリン 及びペグインターフェロンα との併用で治療に用いるために開発している。本薬の臨床での予定 投与方法は300 mg(150 mg×2)1日2回投与である。 バニプレビルは生物薬剤学分類システム(BCS)でクラス4に分類される薬物で、in vivo での吸 収はその溶解性と透過性により制限される可能性が考えられたため、バニプレビルの製剤開発で はLFC 製剤を選択し、カプセル内の脂質基剤で自己乳化させて薬物を送達するシステム(SEDDs) を採用した。臨床開発における主要な製剤はすべてLFC 製剤であり、その概略を以下に示す。 1) 開発初期製剤(FFP 製剤): バニプレビル を油系溶媒:界面活性剤( : )混合液に溶解させた液をカ プセルに充てんした製剤。 2) プロトタイプの市販候補製剤(pPMF 製剤): 使用する添加剤の総量を減らすため、FFP 製剤から、カプセルに充てんする薬液の溶媒組 成を変更して製造した製剤。なお、本製剤では、 を上げるため、バニプレビ ル を用いた。 3) バニプレビル を用いた市販候補製剤〔PMF( )製剤〕: pPMF 製剤で使用していた硬ゼラチンカプセルから、より薬液の充てん容量を大きくする ために軟ゼラチンカプセルに変更した製剤。 4) バニプレビル を用いたPMF 製剤〔PMF( )製剤〕: 製造工程の堅牢性や を向上させるため、原薬をバニプレビル に変更した製剤。 5) 最終製剤(FMI 製剤): 市販予定製剤。薬液の処方は PMF( )製剤と同一であり、PMF( )製剤と の相違点は軟ゼラチンカプセル中の着色剤の添加量がわずかに少ないことのみである。 これら製剤の崩壊、溶出、経口バイオアベイラビリティ及び食事の影響等のバニプレビルの生 物薬剤学的な特性は、in vitro 及び in vivo 試験で検討し、以下の結論を得た。 1) 重要な第Ⅲ相試験を含む臨床試験で用いたバニプレビルの主要なLFC 製剤及び FMI 製剤 間のin vivo 挙動の類似性又は同等性が示された。 2) In vitro 試験で得た崩壊及び溶出データより、pPMF 製剤、PMF( )製剤、PMF(
)製剤及びFMI 製剤の軽微な変更に関して、in vivo 挙動の類似性が示唆された。また、
in vitro 溶出データより PMF( )製剤、PMF( )製剤及びFMI 製剤が生物学 的に同等であることが示された。よって、製剤間の関係を確認するためのヒト生物学的同 等性試験は必要ない。
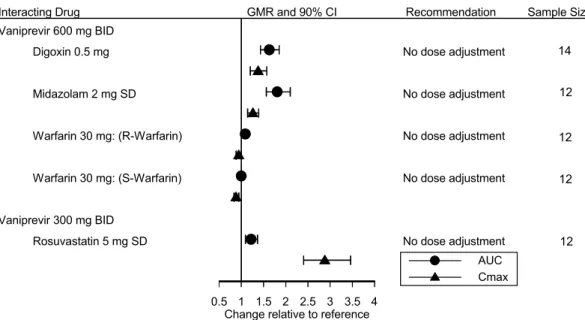
食後投与によりバニプレビルの血漿中曝露量はやや増加したが、臨床的に意味のある変化では なかった。すなわち、バニプレビル PMF( )製剤を、臨床推奨用量の300 mg で標準和朝 食後に日本人の健康被験者に投与したところ、空腹時投与に比べAUC0-∞は34%、Cmaxは47%増大 した。摂食によるTmaxへの影響は認められなかった。観測された薬物動態変化は臨床的に有意な 変化であるかどうかを判断するために設定した変動許容区間の範囲内であり、食事によるバニプ レビルの薬物動態変化は臨床的に意味はないと考えられた。バニプレビルを単剤で経口投与する 際、バニプレビルは食事の制限なく投与可能であると考えられる。 2.7.1.1.1 製剤開発の概観 原薬であるバニプレビルフリー体無水物は白色の粉末で、わずかに吸湿性を有する結晶性粉末 である。バニプレビルの分子式はC38H55N5O9S、分子量は757.94である。バニプレビルの化学構造 を[図2.7.1: 2]に示す。バニプレビルフリー体の飽和水溶液の pH は6.7、電位差滴定による pKa は 5.50、log D(pH7)は4.12であった。 [資料 3.2.S.1.2] 図2.7.1: 2 バニプレビル(MK-7009、 )の化学構造 臨床試験で使用した主要な製剤を[表 2.7.1: 1]に、これらの製剤の処方を[表 2.7.1: 2]にまとめる。 また、バイオアベイラビリティ比較試験では、主要な4 製剤〔FFP 製剤、pPMF 製剤、PMF( )製剤及びPMF( )製剤〕以外に、4 つの LFC 製剤(nLFC1 製剤、nLFC2 製剤、nLFC4 製剤及びnLFC5 製剤)及び 2 つのフィルムコーティング錠(STD FCT 製剤及び ARG40 FCT 製剤) を評価した。これらの製剤は第Ⅱ相及び第Ⅲ相試験では使用されず、得られた結果は本項の表中 に示す。また、詳細な製剤情報は[資料 3.2.P.2.2]に記載している。PMF 製剤のゼラチンカプセル の代表的な組成の詳細、バイオアベイラビリティ評価試験で使用した静脈内投与製剤など、他の
臨床試験製剤の詳細についても[資料 3.2.P.2.2]を参照のこと。 表2.7.1: 1 バニプレビル臨床開発における主要な製剤 製剤 原薬 薬物含有率 (%) 溶媒‡ 組成比 カプセル タイプ 含量 (mg) 試験番号 FFP 製剤 硬カプセル 5 25 100 本邦第Ⅰ相試験:008, 013, 021 海外第Ⅰ相試験:001, 002, 003, 004, 005, 006, 010, 012, 014, 015, 020, 024, 025, 026, 027, 030, 035, MK-3281 006 海外第Ⅱ相試験:007 pPMF 製剤 硬カプセル 100 本邦第Ⅰ相試験:021 海外第Ⅰ相試験:014 本邦第Ⅱ相試験:016 海外第Ⅱ相試験:009 PMF ( ) 製剤 軟カプセル 150 本邦第Ⅰ相試験:046 海外第Ⅰ相試験:027, 032, 029, 048 本邦第Ⅲ相試験: 043, 044, 045 PMF ( ) 製剤 軟カプセル 150 本邦第Ⅰ相試験:011, 034, 049 海外第Ⅰ相試験:051 ‡ : : の混合比 表2.7.1: 2 バニプレビルの臨床試験で使用した主要製剤の組成 成分 配合目的 1カプセル中の量(mg) FFP§ 製剤 pPMF 製剤 PMF ( ) 製剤 PMF ( ) 製剤 FMI 製剤 バニプレビル † 有効成分 100.0 - - - - バニプレビル † 有効成分 - 100.0 150.0 - - バニプレビル † 有効成分 - - - 150.0 150.0 グリセリン脂肪酸エステル ポリソルベート 80 ポリオキシル 35ヒマシ油 ブチルヒドロキシアニソール ジブチルヒドロキシトルエン 薬液の目標質量 硬ゼラチンカプセル (サイズ00、乳白色) 硬カプセル 118.0‡ 118.0‡ - - - ゼラチン バンドシール 適量 適量 - - - ポリソルベート80 バンドシール 適量 適量 - - - 軟ゼラチンカプセル ( 、 軟カプセル - - ‡ ‡ - 軟ゼラチンカプセル ( 、 ) 軟カプセル - - - - ‡ カプセルの目標総質量 ‡ ‡ ‡ ‡ ‡ † 1カプセル中の有効成分の量はフリー対 に換算、‡ 剤皮及びカプセル総質量は概算、§ FFP では有効成分を5及び25 mg を含有する製剤も使用 [資料 3.2.P.2.2]
2.7.1.1.1.1 開発初期の製剤 臨床試験を開始するためにFFP 製剤を開発した。本製剤は、バニプレビル を : ( : )混合液に溶解した液を、硬ゼラチンカプセル に充てんした製剤である。 を抑制するため、充てんした薬液には少量の 及び を加えている。FFP 製 剤中のバニプレビル(フリー体として)の含有量は5、25及び100 mg であり、最大含有量は有効 成分の溶解度及び硬ゼラチンカプセルの充てん量により制限された。 2.7.1.1.1.2 開発後期の製剤 後期臨床開発では3つの主要な LFC 製剤〔pPMF 製剤、PMF( )製剤及びPMF( ) 製剤〕[表2.7.1: 1]を使用した。これら3製剤と FMI 製剤の製剤開発上の変更点を以下にまとめる。 また、製剤間の関連性を[図2.7.1: 1]に示す。 pPMF 製剤 pPMF 製剤は1日当たりに投与される添加剤/界面活性剤の総量を減らすために開発した。界面 活性剤の量を減らすため、 を溶媒として加えた。pPMF 製剤では、バイ オアベイラビリティ比較試験(014試験)で FFP 製剤と同様の in vivo 挙動を示した LFC 製剤と同 様の溶媒組成( : : = : : )を用いた[2.7.1.2.1.2 項]。また、 を上げるため、薬液中でバニプレビル よりも を示すバニプレビル を原薬として用いた。 PMF( )製剤 PMF( )製剤は第Ⅲ相試験開始前に開発した。薬液充てん量を増やし、カプセルの頑健 性を改善するために、PMF 製剤では、pPMF 製剤に使用した硬ゼラチンカプセルではなく、軟ゼ ラチンカプセルを使用した。150 mg PMF( )製剤と100 mg pPMF 製剤のカプセルの内容物 の成分及び組成比は同じである。 PMF( )製剤 PMF( )製剤は、 を改善し、製造工程の堅牢性を向上させるため、原薬を バニプレビル からバニプレビル に変更した製剤である。 FMI 製剤 150 mg FMI 製剤は市販予定製剤であり、軟ゼラチンカプセルの着色剤の添加量をわずかに変更 させた製剤であり、着色剤を除きPMF( )製剤と同一組成の製剤である。PMF 製剤との溶 出挙動の同等性が示されていることから、FMI 製剤の軽微な変更は in vivo 挙動に影響しないと考 えられる。
2.7.1.1.1.3 バニプレビルの溶解性及びバニプレビル製剤の崩壊及び溶出試験の概略 バニプレビルはBCS クラス4に分類される薬物(低溶解性及び低膜透過性)で、以下に示すデ ータより、その溶解性と膜透過性によりin vivo での吸収が制限されることが予想された。 バニプレビルLFC 製剤の崩壊及び溶出試験方法や使用機器の概略を[表2.7.1: 3]に示す。 表2.7.1: 3 溶出試験方法と規格 製剤 FFP 製剤 pPMF 製剤 PMF( )製剤 PMF( )製剤 FMI 製剤 崩壊性 評価 評価 評価 評価 評価 溶出性 評価せず 評価せず 評価 評価 評価 溶出試験液 - 溶出試験液量 900 mL 900 mL 900 mL 試験条件 パドル パドル パドル 崩壊試験液 水 水 水 水 水 溶出性に関する 規格 - - 崩壊性に関する 規格 LFC 製剤では、薬液中でバニプレビル原薬は完全に溶解しているため、pH7付近での LFC 製剤 の溶出挙動は主にカプセルの崩壊によって決まる。臨床試験で使用した製剤の主要なロットにつ いて測定した崩壊時間(カプセル破裂までの時間)の結果を[表2.7.1: 4]にまとめた。これら製剤 のカプセルは速やかに崩壊しており( 分以下)、崩壊特性は許容可能であることが確認された。 また、PMF 製剤の代表的なロットの溶出特性を[資料3.2.P.5.4]に示す。平均溶出率は 分間で % を上回っており、許容基準を満たしていた。PMF( 及び )製剤とFMI 製剤の pH での溶出挙動を[図2.7.1: 3]に示す。平均溶出率及び個々の溶出率の結果から、PMF 製剤及び FMI 製剤の溶出挙動の同等性が示された。すなわち、平均溶出率の比較では、f2関数の値は PMF( )製剤と PMF( )製剤で 、PMF( )製剤と FMI 製剤で 、PMF( ) 製剤と FMI 製剤で であった。また、 分の時点で標準製剤である PMF( )製剤の平均 溶出率は %を超えており、その際の試験製剤の個々の溶出率は平均溶出率の± %以内であっ た。 PMF( )製剤の溶出挙動を3つの異なる pH( 、 及び )の試験液で評価した結果、 pH 及びpH では 分までにほぼ完全に溶出したが、pH では試験時間中の薬物の溶出率は %未満であった[資料3.2.P.2.3項]。pH では ものの、 は認められなかった 、 であると考 えられる。しかしながら、 影響しないと推定される。したがって、
は、in vivo で下部消化管におけるバニプレビルの溶出には影響しないと推察される。 表2.7.1: 4 臨床試験使用製剤の主要バッチにおける崩壊時間 バッチ番号 製剤 崩壊条件 平均崩壊時間(範囲) (min) WL00023354 5 mg FFP 製剤 水、 n=6 WL00025550 100 mg FFP 製剤 水、 n=6 WL00025551 100 mg FFP 製剤 水、 n=6 WL00025552 25 mg FFP 製剤 水、 n=6 WL00029579 100 mg FFP 製剤 水、 n=6 WL00030678 100 mg pPMF 製剤 水、 n=6 WL00030680 100 mg pPMF 製剤 水、 n=6 WL00033332 100 mg pPMF 製剤 水、 n=6 WL00033773 100 mg pPMF 製剤 水、 n=6 WL00034194 150 mg PMF( )製剤 水、 n=6 WL00042510 150 mg PMF( )製剤 水、 n=6 WL00048022 150 mg FMI 製剤 水、 n=6 WL00048007 150 mg FMI 製剤 水、 n=6 [資料 3.2.P.5.4]. [資料 3.2.P.8.3] [資料 3.2.P.2.2] [資料 3.2.P.2.2] 図2.7.1: 3 PMF( )製剤、PMF( )製剤及びFMI 製剤の溶出挙動の比較 溶出試験条件:パドル法 N=12
バニプレビルフリー体の溶解性を水及び数種類の有機溶媒で検討し、[表2.7.1: 5]に示す結果を 得た(詳細は[資料3.2.S.1.3])。水溶性(溶解度: mg/mL)は低いと考えられる。見かけの膜 透過係数は cm/sec [3.2.P.2.2.3 項]であった。これらの結果から、バニプレビル はBCS でクラス4に分類される化合物であり(低溶解性及び低膜透過性)、in vivo でのバニプレビ ルの吸収は溶解性と膜透過性に制限される可能性がある。 表2.7.1: 5 バニプレビルフリー体の溶解性 溶媒 溶解度(mg/mL) 溶解性 水 極めて溶けにくい トルエン 酢酸2-プロピル メタノール ヘプタン アセトニトリル N, N ジメチルアセトアミド エタノール(99.5) > [資料 3.2.S.1.3] 2.7.1.1.2 バニプレビル製剤のin vivo での挙動に関するまとめ In vivo データの解析結果や in vitro の崩壊及び溶出データ全体を通じ、臨床開発上、重要な臨床 試験(第Ⅲ相試験を含む)に使用した製剤と市販予定製剤のin vivo 挙動は類似していると考えら れる[2.7.1.3.1 項]。臨床開発における主要な製剤はすべて LFC 製剤であり、これら製剤間の関連 性を確認するために使用したデータの概要を[図2.7.1: 1]に示した。 臨床開発初期の製剤の生物薬剤学的特性を直接評価した臨床試験2試験(非日本人健康成人男女 を対象とした014試験及び日本人健康成人男性を対象とした021試験)で、pPMF 製剤投与時の曝 露 量 は 同 用 量 の FFP 製 剤 投 与 時 と 概 し て 同 程 度 で あ る こ と が 示 さ れ た [2.7.1.2.1.2 項 ] [2.7.1.2.1.3 項]。 臨床試験で直接的にpPMF 製剤、PMF( )製剤及びPMF( )製剤の相対バイオア ベイラビリティを評価していないが、併合臨床試験データによる統合薬物動態解析では、これら LFC 製剤の違いはバニプレビルの薬物動態に対して有意な影響因子ではなく、FFP 製剤から PMF ( )製剤に亘る製剤で、in vivo 挙動の類似性が支持された[2.7.1.3.1 項]。 バニプレビル原薬は内容液に溶解しているので、内溶液の組成比を変えていないpPMF 製剤か らFMI 製剤までの製剤間の関連性は in vitro の崩壊及び溶出データにより確認した。崩壊時間は これらLFC 製剤すべてで 分以下であり[2.7.1.1.1.3 項]、臨床試験で認められた Tmax(1.0~3.0時 間)よりも短かった[2.7.2.5 項]。この製剤間の崩壊時間の若干の差によるバニプレビルの吸収へ の影響はないと考えられる。また、PMF( )製剤と PMF( )製剤、PMF( ) 製剤とFMI 製剤、及び PMF( )製剤と FMI 製剤の各製剤間で、f2関数及び個々の溶出率
の値に基づき溶出挙動の同等性が示された [2.7.1.1.1.3 項]。FMI 製剤は内用液の組成は PMF(
)製剤と同一で、軟ゼラチンカプセルの着色剤添加量のわずかな減量は、「経口固形製剤の処
方変更の生物学的同等性試験ガイドライン」(薬食審査発0229第10号2012年2月29日)に記載され
たA 水準相当と判断した。以上、FMI 製剤は臨床試験で使用していないが、in vivo 及び in vitro
データより臨床試験で使用したバニプレビルの主要LFC 製剤と FMI 製剤間の in vivo 挙動の類似 性が支持されたことから、ヒト生物学的同等性試験の実施は必要ないと考えた。 バニプレビル LFC 製剤でのバイオアベイラビリティへの食事の影響については[2.7.1.3.2 項]に 記載する。 2.7.1.1.3 ヒト生体試料中の薬物濃度測定法 バニプレビルのヒト生体試料中(血漿中及び尿中)の薬物濃度測定法の概要を[表2.7.1: 6]にま とめた。[表2.7.1: 6]には測定法ごとにバリデーション実施時の複数検量線検体による日内変動、 及び臨床試験の検体測定時の並行品質保証(QC)検体による日間変動の代表的な結果(分析精度 及び真度)を示した。生体試料中のバニプレビル濃度は、血漿検体は液-液抽出による前処理後、 尿検体は希釈液を直接注入して、液体クロマトグラフィー-タンデムマススペクトロメトリ- (LC-MS/MS)法で測定した。いずれの測定法も各測定サイトの標準操作手順書(SOP)の基準 を満たした。また、すべての測定法でヒト試料中のバニプレビルは許容可能な選択性と特異性を 示した。よって、これら測定法はすべてバニプレビルの臨床検体測定に適応可能と判断した。バ リデーション各項目の検討結果や試料の安定性に関する結果、及び臨床試験でバニプレビルと併 用した薬物の濃度測定法の詳細は各臨床試験総括報告書に付したバリデーション試験報告書、あ るいは検体濃度測定に関する報告書の中で記載している。
表2.7.1: 6 ヒト生体試料中のバニプレビル濃度測定法 測定法 適用した 臨床試験 検体 種類 測定対象 上段:検量線 濃度範囲† 下段:QC 濃度‡ (ng/mL) 上段:日内変動結果§ 下段:日間変動結果|| N (%)真度 分析精度 (%) DM-850 001、002、003、004 血漿 バニプレビル 1~1000 5 -2.1~2.5 1.3~8.6 3, 75, 750 48 -6.7~-3.2 3.6~4.3 DM-850B 004、005、006、006¶、 007、008、009、010、 012、013、014、020、 021、024、027、029††、 035、048†† 血漿 バニプレビル 1~1000 6 -4.0~10.0 2.54~10.0 3, 75, 750 30 -4.3~-1.9 4.0~4.3 DM-938# 015 血漿 バニプレビル 0.025~5 6 -3.2~3.0 2.0~9.2 0.08, 0.3, 3.75 22 0.0~1.1 0.0~3.7 2H 8 -バニプレビル 0.025~5 6 -5.9~2.6 1.7~4.8 0.08, 0.3, 3.75 22 0.0~4.0 3.2~12.5 479 026 血漿 バニプレビル 0.2~100 6 -4.3~2.3 5.5~8.7 0.6, 10, 75 20 -3.1~-1.2 4.6~7.4 479.1 011、016、030、032、 034、046、049、051 血漿 バニプレビル 1~1000 6 -2.41~1.84 1.51~4.86 3, 75, 750 66 -4.7~3.2 3.9~7.3 DM-852 001、008 尿 バニプレビル 200~100000 5 -4.0~3.6 0.8~3.7 600, 10000, 75000 4 -3.0~6.9 4.3~9.1 † 検量線濃度範囲は定量範囲と同じ、‡QC 濃度:品質保証検体濃度、§ 複数の検量線検体(n=5~6)による代表的 な日内変動結果、|| 複数のQC 検体による代表的な日間変動結果(太斜字で示した臨床試験で得られた結果)、 ¶MK-3281の開発試験として実施した試験、# バニプレビル非標識体及び重水素標識体の同時定量法、†† 029試験及 び048試験で実施された肝生検検体はバリデートされていない測定法で検体中バニプレビル濃度を測定した。 測定法DM-850、DM-850B、DM-938及び DM-852は社内(Merck in West Point, PA.)で、測定法479及び479.1は
, で使用した。 [資料5.3.1.4.1: Validation] 2.7.1.2 個々の試験結果の要約 バニプレビルの生物薬剤学的評価を行った臨床薬理試験の概要を[付録2.7.1: 2]にまとめ、これ らの試験の薬物動態の結果を[付録2.7.1: 3]に示す。これらの試験で評価したバニプレビルの用量 は100、300及び600 mg であり、第Ⅱ/Ⅲ相試験で評価した用量をおおむね含む検討結果が得られ た。バニプレビルの薬物動態に及ぼす食事の影響の予備的評価は、用量漸増単回投与薬物動態試 験(001試験)の一部としても実施しており、この結果は[2.7.2.2.2.1 項]で要約する。なお、食事 の影響試験で用いた標準的な高脂肪洋朝食は、米国食品医薬品局(FDA)の「Food-Effect
Bioavailability and Fed Bioequivalence Studies(2002)」で定義されたものである。
2.7.1.2.1 バイオアベイラビリティ比較試験
非日本人を対象に実施した006及び032試験で評価した被験製剤は、その後の臨床開発で更なる 検討を実施しなかった。これら試験については、対照製剤で評価した薬物動態への食事の影響は
要約するが、製剤間比較については記述しない。006及び032試験の製剤間比較の結果は [付録
2.7.1.2.1.1 非日本人健康成人男女を対象としたバイオアベイラビリティ比較試験(FFP 製剤 vs. 固形製剤)(006試験) 非日本人健康成人男女(年齢範囲18~45歳)を対象とした、2種類のフィルムコーティング錠 (STD FCT 製剤及び ARG40 FCT 製剤)(被験製剤)と FFP 製剤(対照製剤)の経口バイオアベ イラビリティを比較評価するための、無作為化、非盲検、2パート、複数パネル、複数投与期、一 部投与順固定、クロスオーバー試験を実施した。本試験のパートⅠでは、100 mg(13例)又は600 mg (15例)の2用量でバニプレビルの薬物動態の製剤間比較及び被験製剤の食事の影響について検討 した。パートⅠの各投与期では被験者に被験製剤又は対照製剤を空腹時(一晩絶食)又は食後(標 準高脂肪洋朝食の摂取開始より30分後)単回経口投与した。パートⅡでは、各投与期で被験者(12 例)にFFP 製剤を600 mg の用量で空腹時(一晩絶食)又は食後(標準高脂肪洋朝食の摂取開始よ り30分後)単回経口投与し、食事の影響を検討した[資料5.3.1.1.1: P006]。 結果及び結論 2種類のフィルムコーティング錠投与時の AUC0-∞及びCmaxは、同一用量のFFP 製剤投与時より 低かった[付録2.7.1: 3]。そのため、これ以降、これらフィルムコーティング錠の評価は実施しな かった。バニプレビル600 mg(FFP 製剤)を食後投与した際、バニプレビルの AUC0-∞の幾何平均 比(GMR)(食後投与/空腹時投与)(90% CI)は0.86(0.71, 1.04)であり、摂食による統計的に 有意な変化はみられなかった。Cmaxは0.61(0.43, 0.89)で摂食による低下が認められた。Tmax(中 央値)は空腹時投与で2.5時間、食後投与で3.0時間であった。 2.7.1.2.1.2 非日本人健康成人男女を対象としたバイオアベイラビリティ比較試験(FFP 製剤 vs. 新規 LFC 製剤)(014試験) 非日本人の健康成人男女(年齢範囲21~45歳)を対象とした無作為化、非盲検、2パネル、7期 クロスオーバー試験を実施し、空腹時条件下で3種類の新規 LFC 製剤(nLFC1製剤、nLFC2製剤及 びpPMF 製剤)(被験製剤)についてバニプレビルの経口バイオアベイラビリティをFFP 製剤(対 照製剤)と比較した。また、これら3種類の被験製剤を投与した際のバニプレビルの薬物動態に対 する食事の影響を検討した。検討は100 mg(12例)及び600 mg(12例)の2用量で実施した。各 投与期で被験者にバニプレビルを被験製剤又は対照製剤で、空腹時(一晩絶食)又は食後(標準 高脂肪洋朝食の摂取開始より30分後)に単回経口投与した[資料5.3.1.2.1: P014]。 結果及び結論 3種類の LFC 製剤を空腹時投与した際の AUC0-∞及びCmaxは、同一用量のFFP 製剤を空腹時投 与した際と類似していた。このうちpPMF 製剤は、100及び600 mg の用量のいずれにおいても FFP 製剤に最も類似しており、100 mg 投与時の GMR(pPMF 製剤/FFP 製剤)(90% CI)は AUC0-∞
で0.99(0.90, 1.09)、Cmaxで1.29(1.08, 1.55)、600 mg 投与時は AUC0-∞で0.90(0.66, 1.23)、Cmax
で0.76(0.49, 1.16)であった。この結果に基づき、その後の臨床開発には pPMF 製剤を使用した。
空腹時)(90% CI)]は、100 mg 投与時の AUC0-∞で0.84(0.69, 1.02)、Cmaxで0.36(0.17, 0.77)、600 mg 投与時のAUC0-∞で1.32(0.95, 1.84)、Cmaxで1.14(0.72, 1.81)であった。また、100及び600 mg 投 与時のTmax(中央値)は空腹時でそれぞれ1.0及び2.5時間、食後でそれぞれ1.5及び3.0時間であっ た。 2.7.1.2.1.3 日本人健康成人男性を対象としたバイオアベイラビリティ比較試験(FFP 製剤 vs. pPMF 製剤)(021試験) 日本人健康成人男性(年齢範囲21~44歳)を対象に無作為化、非盲検、2パート、2パネル、2 期、クロスオーバー試験を実施し、pPMF 製剤(被験製剤)投与時のバニプレビルの経口バイオ アベイラビリティをFFP 製剤(対照製剤)投与時と比較評価した。また、pPMF 製剤100 mg 単回 経口投与時のバニプレビルの薬物動態を検討した。パートⅠは、300 mg(12例)又は600 mg(11 例)の2用量で検討を実施し、各投与期で被験者に、pPMF 製剤又は FFP 製剤を用いて空腹時(一 晩絶食後)にバニプレビルを単回経口投与した。パートⅡではpPMF 製剤を100 mg の用量で被験 者にバニプレビルを単回経口投与した[資料5.3.1.2.2: P021]。パートⅡの薬物動態の結果及び考察 は[付録2.7.1: 3]及び [2.7.2.3.1 項]を参照のこと。 結果及び結論 pPMF 製剤投与時の AUC0-∞及びCmaxは、300及び600 mg のいずれの用量においても FFP 製剤投 与時と類似していた。pPMF 製剤と FFP 製剤を比較した AUC0-∞のGMR(pPMF 製剤/FFP 製剤)
(90% CI)は300 mg で1.00(0.80, 1.23)、600 mg で0.81(0.64, 1.01)であり、CmaxのGMR(90% CI)
は、300 mg で1.22(0.85, 1.74)、600 mg で0.78(0.59, 1.05)であった。Tmaxにはこれらの製剤間で 統計学的な有意差は認められなかった。 2.7.1.2.1.4 非日本人健康成人男女を対象としたバイオアベイラビリティ比較試験〔PMF( )製剤vs. 新規 LFC 製剤〕(032試験) 非日本人健康成人男女(年齢範囲27~45歳)を対象に無作為化、非盲検、2パート、複数投与期、 クロスオーバー試験を実施し、2種類の新規 LFC 製剤(nLFC4製剤及び nLFC5製剤)(被験製剤) 投与時のバニプレビルの経口バイオアベイラビリティを300 mg(16例)及び/又は600 mg(18例) の2用量で PMF( )製剤(対照製剤)投与時と比較評価した。各投与期で被験者にバニプ レビルを LFC 製剤又は PMF( )製剤で、空腹時(一晩絶食)又は食後(標準高脂肪洋朝 食の摂取開始より30分後)に単回経口投与した[資料5.3.1.2.3: P032]。 結果及び結論 2種類の LFC 製剤投与時の AUC0-∞及びCmaxは、同一用量のPMF( )製剤投与時と類似 していることが示された[付録2.7.1: 3]。なお、これ以降、nLFC4製剤及び nLFC5製剤を用いた検 討は実施しなかった。 600 mg の用量では、食事摂取により PMF( )製剤投与時のAUC0-∞及びCmaxは増加し、
これら薬物動態パラメータのGMR(食後/空腹時)(90% CI)は、それぞれ1.49(1.19, 1.87)及 び1.45(1.04, 2.01)であった。300 mg の用量では、AUC0-∞のGMR(食後/空腹時)(90% CI)は 1.25(1.08, 1.45)で、食事摂取により AUC0-∞が増加したが、Cmaxは0.96(0.69, 1.34)で統計学的 に有意な影響はみられなかった。Tmax(中央値)には、いずれの用量でも食事の影響は認められ なかった。 なお、nLFC4製剤及び nLFC5製剤に関する食事の影響の結果は[付録2.7.1: 3]を参照の こと。 2.7.1.2.2 バイオアベイラビリティ及び食事の影響に関する試験 2.7.1.2.2.1 バニプレビルの絶対バイオアベイラビリティ評価試験(015試験) 非日本人健康成人男女(年齢範囲32~76歳)を対象に、バニプレビルの絶対経口バイオアベイ ラビリティの推定を目的とした一部盲検、無作為化、プラセボ対照、3期、一部投与順固定、クロ スオーバー試験を実施した。第1期及び第2期では、10例の被験者にバニプレビル非標識体(FFP 製剤)を100又は600 mg の用量で単回経口投与し、その1.75時間後にバニプレビル重水素標識体 を10 mg の用量で静脈内投与した。本試験では、静脈内投与されたバニプレビル重水素標識体と 経口投与されたバニプレビル非標識体の血漿中濃度はそれぞれ区別して測定した。この評価法で は静脈内用量及び経口用量から得られた曝露量を同じ非線形条件で評価することができるため、 バニプレビルのように非線形性の薬物動態を示す化合物であっても、種々の経口用量でバイオア ベイラビリティを算出することができる。第3期では、被験者にバニプレビル重水素標識体10 mg と非標識体10 mg をともに単回静脈内投与で併用し、重水素標識によるバニプレビル薬物動態へ の影響を検討した[資料5.3.3.4.2: P015]。 結果及び結論 バニプレビルの重水素標識化はバニプレビルの薬物動態に影響しなかった[付録2.7.1: 3]。バニ プレビルを100及び600 mg の用量で経口投与した際のバイオアベイラビリティは、それぞれ7.9% 及び33.0%であった[表2.7.1: 7]。バニプレビル重水素標識体10 mg の静脈内投与にバニプレビル非 標識体100 mg の経口投与、あるいは非標識体10 mg の静脈内投与を併用投与した際、重水素標識 体濃度に基づいて算出したバニプレビルの血漿クリアランス(CLp)及び定常状態分布容積(Vdss) は類似していた(非標識体100 mg の経口投与併用時:約38 L/hr 及び約42 L、非標識体10 mg 静脈 内投与併用時:約40 L/hr 及び約48 L)。非標識体600 mg の経口投与併用時では CLpは約24 L/hr、 Vdssは約24 L に減少した。
表2.7.1: 7 バニプレビル非標識体(経口投与)及び重水素標識体(静脈内投与)を併用した際 の血漿中薬物動態パラメータの要約統計量(015 試験) 処方A:100 mg 経口(非標識) 処方B :600 mg 経口(非標識) 薬物動態パラメータ N 幾何平均 95% CI N 幾何平均 95% CI F†¶ (%) 9 7.86 (6.29, 9.83)‡ 10 33.0 (26.8, 40.7)‡ AUC0-∞†(μM•hr) 9 0.274 (0.190, 0.393) 10 10.9 (7.72, 15.5) Cmax †(μM) 10 0.125 (0.089, 0.176) 10 4.92 (3.49, 6.95) Tmax §(hr) 10 1.00 (1.00, 1.98) 10 2.75 (1.72, 3.08) t½ || (hr) 9 6.26 (25) 10 4.00 (19) 処方A:10 mg 静脈内(標識) 処方B:10 mg 静脈内(標識) 薬物動態パラメータ N 幾何平均 95% CI N 幾何平均 95% CI AUC0-∞†(μM•hr) 10 0.340 (0.283, 0.408) 10 0.546 (0.455, 0.655) Ceoi †(μM) 10 1.18 (0.974, 1.42) 10 1.22 (1.01, 1.47) CLp†(L/hr) 10 38.4 (32.0, 46.1) 10 23.9 (19.9, 28.7) Vdss †(L) 10 41.9 (34.4, 50.9) 10 24.5 (20.1, 29.8) t½ ||(hr) 10 5.24 (24) 10 3.89 (16) Ceoi:投与終了時の血漿中濃度、CLp:血漿クリアランス、Vdss:定常状態分布容積 †対数変換値に基づく混合効果モデルで算出した逆変換最小二乗平均及びCI、‡90% CI、§ 中央値(最小値, 最大 値)、||幾何平均(幾何変動係数%)、¶ 絶対経口バイオアベイラビリティ(%)= 100 ×{(AUC 0-∞, PO×DoseIV ) /(AUC0-∞, IV×DosePO )×0.9895 } [資料5.3.3.4.2: P015] 2.7.1.2.2.2 日本人健康成人男女を対象とした食事の影響試験(049試験) 日本人健康成人男女(年齢範囲21~35歳)を対象とした、非盲検、無作為化、2期、クロスオー バー試験を実施し、バニプレビル FMI 製剤と同等な PMF( )製剤を用いて単回経口投与 後の薬物動態に及ぼす食事の影響を評価した。各投与期で被験者にPMF( )製剤を用いて バニプレビル300 mg を空腹時(一晩絶食)又は食後(標準的和朝食の摂取開始より30分後)に単 回経口投与した[資料5.3.1.1.2: P049]。 結果及び結論 バニプレビルの空腹時及び食後投与時の薬物動態パラメータを比較した[表2.7.1: 8]。本試験で
得られたAUC0-∞及びCmaxのGMR(食後/空腹時)[90% 信頼区間(CI)]はそれぞれ1.34(1.13,
1.58)及び1.47(1.15, 1.89)であり、摂食により AUC0-∞及びCmaxは増加した。Tmax(中央値)に
表2.7.1: 8 日本人健康成人被験者にバニプレビル 300 mg を PMF( )製剤で空腹時あるいは食後単回経口投与 した際の平均血漿中薬物動態パラメータの要約統計量(049 試験) 薬物動態 パラメータ 食後 空腹時 食後/空腹時 個体内 変動係数 (%) N GM 95% CI N GM 95% CI GMR 90% CI AUC0-12 hr(μM∙hr) 14† 2.39 (1.72, 3.33) 15 1.78 (1.37, 2.31) 1.34 (1.13, 1.60) 26 AUC0-∞†(μM∙hr) 14 2.53 (1.83, 3.49) 15 1.89 (1.47, 2.44) 1.34 (1.13, 1.58) 25 Cmax†(nM) 14 1032 (651, 1636) 15 701 (511, 960) 1.47 (1.15, 1.89) 37 C12 hr†(nM) 14 14.5 (12.2, 17.3) 15 13.5 (10.9, 16.6) 1.08 (0.95, 1.22) 19 Tmax‡(hr) 14 2.00 (1.00, 4.00) 15 2.00 (1.00, 6.00) - - - t1/2§(hr) 14 6.55 (35) 15 6.15 (29) - - - GM:幾何平均、-:該当無し †対数変換値に基づく混合効果モデルで算出した逆変換最小二乗平均及びCI、‡中央値(最小値, 最大値)、 §幾何平均(幾何変動係数%) [資料5.3.1.1.2: P049] 2.7.1.3 全試験を通しての結果の比較と解析 バニプレビルの製剤間での薬物動態比較は、in vitro の崩壊及び溶出試験結果、個々のバイオア ベイラビリティ比較試験の結果、及び複数の臨床試験のノンコンパートメント解析結果を併合し て実施した統合薬物動態解析の結果に基づいて行った[資料5.3.3.3.2: COMP]。 バニプレビルの薬物動態に対する食事の影響は、日本人健康成人男女を対象にFMI 製剤と生物 学的に同等な PMF( )製剤を用いて実施した食事の影響試験(049試験)の結果に基づい て評価した。また、複数の臨床試験のノンコンパートメント解析結果を併合して実施した統合薬 物動態解析でも評価した[資料5.3.3.3.2: COMP]。 2.7.1.3.1 バイオアベイラビリティ バニプレビルの絶対経口バイオアベイラビリティを非日本人被験者を対象に評価した。バニプレビ ルの絶対バイオアベイラビリティは100 mg 投与時で7.9%(90% CI:6.3~9.8%)、600 mg 投与時で33.0% (90% CI:26.8~40.7%)であり、低度から中等度であった。用量漸増単回経口投与後、バニプレビル の曝露は用量比例性を上回る増大を示した。この用量比例性を上回る曝露増大には肝取り込みの飽和 や初回通過による消失の阻害が主として起因しており[2.7.2.3.1 項]、バニプレビルの溶解性や膜透過性 による吸収制限には関連していないと考えられる。 臨床開発に使用したバニプレビルの4つの主要な LFC 製剤のうち、大部分の第Ⅰ相試験で使用 したFFP 製剤及び日本人 HCV 患者を対象とした第Ⅱ相用量設定試験で使用した pPMF 製剤は、 臨床試験でのバイオアベイラビリティ比較により類似性を確認した。また、LFC 製剤は、バニプ レビルの原薬を油系溶媒に溶解させ、その薬液をカプセルに充てんすることにより製造され、い ずれのLFC 製剤もカプセルは速やか( 分以下)に崩壊することが確認されたことから、pPMF 製剤及び重要な第Ⅲ相試験で用いたPMF( )製剤、並びにPMF( )製剤及び薬液組 成がFMI 製剤と同一な PMF( )製剤間のin vivo 挙動は類似していることが示唆された。
さらに、PMF( )製剤とPMF( )製剤、PMF( )製剤とFMI 製剤、及び PMF ( )製剤とFMI 製剤間で算出した f2関数の値及び個々の溶出率から、これら製剤間の溶出 挙動は同等であることが示された[2.7.1.1.1.3 項]。 以上の結果より、重要な第Ⅲ相試験を含むバニプレビルの臨床試験で使用した主要なLFC 製剤 及びFMI 製剤の in vivo 挙動の類似性又は同等性が示された。 FFP 製剤及び pPMF 製剤投与時の血漿中薬物動態を日本人(021試験)及び非日本人(014試験) の健康被験者を対象とした試験で直接比較した。日本人健康被験者では、想定される臨床推奨用 量300 mg で、AUC0-∞のGMR(pPMF 製剤/FFP 製剤)の90% CI は(0.80, 1.25)の範囲(AUC0-t に対し、生物学的同等性の基準とされる範囲。なお、これら試験で AUC0-tは評価していない。) に入り、この2製剤によるバイオアベイラビリティは同程度であることが示された。また、Cmax のGMR(pPMF 製剤/FFP 製剤)の90% CI は1を挟んでおり、製剤間で統計学的有意差は認めら れなかった [表2.7.1: 9]。また、非日本人健康被験者や他の用量(100及び600 mg)でも、pPMF 製剤とFFP 製剤の製剤間で AUC0-∞及びCmaxに概して統計学的有意差はみられなかったが、GMR の90% CI は(0.80, 1.25)の範囲から外れていた。600 mg の用量を投与した際の AUC0-∞及びCmax のGMR の90% CI は、日本人と非日本人健康被験者で同程度であった。以上、FFP 製剤と pPMF 製剤を同用量で投与した際の薬物動態は概して類似していると考えられる。 表2.7.1: 9 非日本人(014 試験)及び日本人(021 試験)健康成人での pPMF 製剤と FFP 製剤の血漿中曝露比較 製剤間比較: pPMF 製剤 vs FFP 製剤 N AUC0-∞ Cmax
GMR(90% CI)† rMSE GMR(90% CI)† rMSE
300 mg、日本人(021試験) 12‡ 1.00(0.80, 1.23) 0.270 1.22(0.85, 1.74) 0.458 600 mg、日本人(021試験) 11§ 0.81(0.64, 1.01) 0.280 0.78(0.59, 1.05) 0.356 100 mg、非日本人(014試験) 12 0.99(0.90, 1.09) 0.135 1.29(1.08, 1.55) 0.261 600 mg、非日本人(014試験) 12 0.90(0.66, 1.23) 0.450 0.76(0.49, 1.16) 0.613 rMSE:線形混合効果モデルで得られた条件付き平均二乗誤差(残差)の平方根。100を乗じると実数尺度での被験者内変動係 数の近似値となる。 †pPMF 製剤/FFP 製剤、‡pPMF 製剤 N=12及び FFP 製剤 N=10、§pPMF 製剤 N=10及び FFP 製剤 N=11 [資料 5.3.1.2.2: P021] [資料 5.3.1.2.1: P014] pPMF 製剤、PMF( )製剤、PMF( )製剤の相対的なバイオアベイラビリティを 直接評価する臨床試験は実施していないが、複数試験の併合データによる多変量統合薬物動態解 析では製剤間の類似性を支持する結果が得られた。本解析の結果、製剤間における薬物動態パラ メータ(AUC0-∞とCmax)のGMR の90% CI は(0.80, 1.25)の範囲から外れており、バニプレビル の血漿中薬物動態にみられる大きなばらつきが影響している可能性が考えられたが、幾何平均は 概して1.0に近い値を示し、100 mg 投与時の Cmaxを除き、バニプレビル主要製剤間で、製剤の違 いはバニプレビルの薬物動態に対し統計学的に有意な共変量ではないことが示された[表2.7.1: 10] [資料5.3.3.3.2: COMP]。
表2.7.1: 10 バニプレビルの薬物動態への製剤の影響に関する統合薬物動態解析結果 用量 (mg) 比較製剤 N † n‡ AUC0-∞ Cmax GMR(90% CI) GMR(90% CI) 100 pPMF 製剤/FFP 製剤 26/42 37/42 1.04(0.87, 1.24) --27/43 38/43 -- 1.33(1.02, 1.74) 300 pPMF 製剤/FFP 製剤 16/66 16/66 1.04(0.78, 1.37) --16/72 16/72 -- 1.30(0.84, 2.01) PMF( )製剤/FFP 製剤 16/66 32/66 0.84(0.69, 1.03) --16/72 32/72 -- 1.03(0.76, 1.41) PMF( )製剤/FFP 製剤 15/66 29/66 1.04(0.79, 1.36) --16/72 30/72 -- 1.11(0.72, 1.70) 600 pPMF 製剤/FFP 製剤 26/90 38/101 1.03(0.86, 1.23) --27/122 39/133 -- 1.00(0.80, 1.24) PMF( )製剤/FFP 製剤 18/90 36/101 0.97(0.79, 1.19) --18/122 36/133 -- 1.09(0.85, 1.39) † 被験者数(食後投与/空腹時投与)、‡データ数(食後投与/空腹時投与) [資料 5.3.3.3.2: COMP] 2.7.1.3.2 食事の影響 バニプレビルの溶解性は低いため、食事はin vivo でのバニプレビルの溶出や吸収に影響する統 合的な要因として、バニプレビルの血漿中薬物動態に影響する可能性がある。バニプレビルの薬 物動態に対する食事の影響は4 つの生物薬剤学試験で検討した。これらの試験結果及び統合薬物 動態解析の結果より、食事はバニプレビルの薬物動態には臨床上問題となる影響を及ぼさず、バ ニプレビルの単独投与は食事の制限なく投与可能であることが示された。 特に、049試験では、臨床開発中及び市販時の食事に関する推奨用法を検討するために、日本人 健康成人被験者に想定される臨床推奨用量300 mg を FMI 製剤と生物学的に同等な PMF( ) 製剤で投与し、標準的和朝食摂取によるバニプレビルのバイオアベイラビリティへの影響を評価
した。その結果、摂食によりAUC0-∞及びCmaxはそれぞれ34%及び47%増加したが、Tmaxには影響
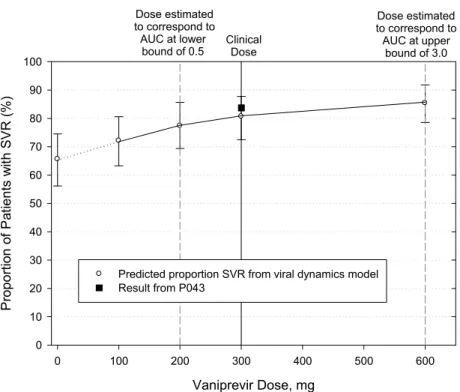
は認められなかった[付録2.7.1: 3]。これらの変化は臨床的に意味のある薬物動態変化(血漿中曝 露変化)を判定するための変動許容区間(0.5, 3.0)[2.7.2.1.3.1.2 項]の範囲内であったため、臨床 的に問題とはならないと考えられた。バニプレビルを単剤で経口投与する際、バニプレビルは食 事の制限なく投与可能であると考えられる。 非日本人健康成人被験者を対象とした3試験(006、014及び032試験)は第Ⅱ相試験の食事に関 する推奨用法を検討するために実施した。 非日本人健康成人を対象に、バニプレビルpPMF 製剤及び PMF( )製剤を標準的な高脂 肪洋朝食後に投与したところ、300 mg 以上の用量(300及び600 mg)で概して AUC0-∞の増加がみ られたが、これらの用量ではCmaxに有意な影響は認められなかった。バニプレビルpPMF 製剤を 100 mg 投与した際は、食事により Cmaxの低下がみられた。 複数試験の併合データによる多変量統合薬物動態解析では、食後投与によりバニプレビルの曝 露量が軽度に増加することが示された。いずれの製剤及び被験者集団でも、300及び600 mg の用 量でのAUC0-∞は食後投与で概して約20~30%増加した。Cmaxには摂食による一貫した有意な影響
は認められなかった [表2.7.1: 12] [資料5.3.3.3.2: COMP]。 バニプレビルに対する食事の影響は、[付録2.7.1: 4]にフォレストプロットとして示す。 食事の影響に対する予備的評価は、用量漸増単回経口投与試験(001試験)の一部として80 mg の用量で実施した。この用量はバニプレビルの臨床開発で一貫して使用されなかったため、[表 2.7.1: 11] に食事の影響の試験結果を含めたが、統合薬物動態解析には含めず、本項でもこれ以上 の議論はしなかった。 表2.7.1: 11 バニプレビルの薬物動態に対する食事の影響 試験番号 (被験者) 製剤 食事内容 投与量 (mg) N AUC 0-∞ (食後/空腹時) C max (食後/空腹時) GMR (90% CI) rMSE GMR (90% CI) rMSE 049 (日本人) PMF ( ) 製剤 和朝食 300 14 1.34 (1.13, 1.58) NA 1.47 (1.15, 1.89) NA 014 (非日本人) pPMF 製剤 高脂肪食 100 11~12 0.84 (0.69, 1.02) 0.260 0.36 (0.17, 0.77) 1.082 014 (非日本人) pPMF 製剤 高脂肪食 600 12 1.32 (0.95, 1.84) 0.450 1.14 (0.72, 1.81) 0.630 032 (非日本人) PMF ( ) 製剤 高脂肪食 300 16 1.25 (1.08, 1.45) 0.249 0.96 (0.69, 1.34) 0.566 032 (非日本人) PMF ( ) 製剤 高脂肪食 600 18 1.49 (1.19, 1.87) 0.407 1.45 (1.04, 2.01) 0.590 001 (非日本人) FFP 製剤 高脂肪食 80 4~6 1.22 (1.02, 1.46) 0.144 0.79 (0.54, 1.17) 0.396 006 (非日本人) FFP 製剤 高脂肪食 600 11 0.86 (0.71, 1.04) 0.243 0.61 (0.43,0.89) 0.473 rMSE:線形混合効果モデルで得られた条件付き平均二乗誤差(残差)の平方根。100を乗じると実数尺度での被験者内変動係 数の近似値となる。 [資料 5.3.1.1.2: P049][資料 5.3.1.2.1: P014] [資料 5.3.1.2.3: P032] [資料 5.3.3.1.1: P001] [資料 5.3.1.1.1: P006]
表2.7.1: 12 バニプレビルの薬物動態への食事の影響に関する統合薬物動態解析結果 投与量 (mg) 食事内容による比較 N † n‡ GMR(90% CI) AUC0-∞ Cmax 100 高脂肪食/空腹時 11/56 11/68 0.83(0.67, 1.03) --11/58 11/70 -- 0.52(0.37, 0.73) 300 和朝食/空腹時 14/103 14/113 1.31(1.03, 1.67) --15/109 15/119 -- 1.46(1.00, 2.12) 高脂肪食/空腹時 16/103 16/113 1.25(0.99, 1.57) --16/109 16/119 -- 0.96(0.67, 1.38) 600 高脂肪食/空腹時 41/112 41/134 1.22(1.02, 1.44) --41/145 41/167 -- 0.95(0.76, 1.17) † 被験者数(食後投与/空腹時投与)、‡データ数(食後投与/空腹時投与) [資料 5.3.3.3.2: COMP]
2.7.1.4 付録 バイオアベイラビリティ比較試験では、主要な4製剤〔FFP 製剤、pPMF 製剤、PMF( ) 製剤及び PMF( )製剤〕以外に、4つの液体充てんカプセル剤(nLFC1製剤、nLFC2製剤、 nLFC4製剤及び nLFC5製剤)、並びに2つのフィルムコーティング錠(STD FCT 製剤及び ARG40 FCT 製剤)を評価した。これらの製剤は第Ⅱ相及び第Ⅲ相試験では使用されず、得られた結果は 本項の表中に示す。また、詳細な製剤情報は[3.2.P.2.2 項]に記載している。 付録2.7.1: 1 バニプレビルの臨床試験で使用した製剤 試験 番号 実施 場所 相 試験内容 製剤 含有量† (mg) 製剤コード (ロット番号) 健康被験者及び特別集団 001 海外 Ⅰ 単回漸増投与試験 FFP 製剤 5 WL00022329 25 WL00022328 100 WL00022330 002 海外 Ⅰ 反復漸増投与試験 FFP 製剤 100 WL00023350 003 海外 Ⅰ 健康高齢男性及び女性による薬物動態 試験 FFP 製剤 5 WL00023354 25 WL00023352 100 WL00023350 005 海外 Ⅰ 肝機能障害患者での薬物動態試験 FFP 製剤 100 WL00033401 006 海外 Ⅰ バイオアベイラビリティ比較試験 FFP 製剤 100 WL00025551 STD FCT 製剤 100 WL00027170 ARG40 FCT 製剤 100 WL00027207 008 国内 Ⅰ 単回漸増投与試験 FFP 製剤 5 WL00023354 25 WL00025552 100 WL00025551 010 海外 Ⅰ ミダゾラムとの薬物相互作用試験 FFP 製剤 100 WL00025551 011 国内 Ⅰ Thorough-QT 試験 PMF( )製剤 150 WL00042510 012 海外 Ⅰ ADME 試験 FFP 製剤 100 WL00029046 013 国内 Ⅰ 反復漸増投与試験 FFP 製剤 100 WL00025550 014 海外 Ⅰ バイオアベイラビリティ比較試験 FFP 製剤 100 WL00029579 nLFC1製剤 100 WL00029737 nLFC2製剤 100 WL00029738 pPMF 製剤 100 WL00029892 015 海外 Ⅰ 絶対バイオアベイラビリティ評価単回 投与試験 FFP 製剤 100 WL00029579 IV 製剤 1 mg/mL WL00030386 1 mg/mL WL00031828 020 海外 Ⅰ ケトコナゾールとの薬物相互作用試験 FFP 製剤 100 WL00029579 021 国内 Ⅰ バイオアベイラビリティ比較試験 FFP 製剤 100 WL00029579 pPMF 製剤 WL00030680 024 海外 Ⅰ ジゴキシンとの薬物相互作用試験 FFP 製剤 100 WL00032992 025 海外 Ⅰ ワルファリンとの薬物相互作用試験 FFP 製剤 100 WL00029579 026 海外 Ⅰ リファンピシンとの薬物相互作用試験 FFP 製剤 100 WL00029579 027 海外 Ⅰ 高用量投与試験 FFP2製剤 150 WL00033015 FFP 製剤 100 WL00032992 PMF( )製剤 150 WL00034194 030 海外 Ⅰ ジルチアゼムとの薬物相互作用試験 FFP 製剤 100 WL00032992 032 海外 Ⅰ バイオアベイラビリティ比較試験 nLFC4製剤 150 WL00034585 nLFC5製剤 150 WL00034584 PMF( )製剤 150 WL00034194 034 国内 Ⅰ フェニトインとの薬物相互作用試験 PMF( )製剤 150 WL00042510 035 海外 Ⅰ 高用量投与試験 FFP 製剤 100 WL00029065
付録2.7.1: 1 バニプレビルの臨床試験で使用した製剤(続き) 試験 番号 実施 場所 相 試験内容 製剤 含有量† (mg) 製剤コード (ロット番号) 健康被験者及び特別集団(続き) 046 国内 Ⅰ ロスバスタチンとの薬物相互作用試験 PMF( )製剤 150 WL00036335 049 国内 Ⅰ 食事の影響試験 PMF( )製剤 150 WL00042510 051 海外 Ⅰ リファンピシンとの薬物相互作用試験 PMF( )製剤 150 WL00042510 006 (MK-3281) 海外 Ⅰ リトナビルとの薬物相互作用試験 FFP 製剤 100 WL00033773 HCV 感染患者 004 海外 Ⅰ HCV 感染患者による安全性・有効性・薬 物動態試験 (バニプレビル単剤) FFP 製剤 5 WL00023354 25 WL00025552 WL00023352 100 WL00023350 WL00025551 007 海外 Ⅱ 未治療慢性HCV 感染患者による安全 性・有効性・薬物動態試験 (バニプレビル+PEG2a+RBV) FFP 製剤 25 WL00029059 100 WL00029061 WL00029062 009 海外 Ⅱ 慢性HCV 感染患者による安全性試験 (バニプレビル+PEG2a+RBV) pPMF 製剤 100 WL00033332 WL00033558 WL00033773 WL00038641 016 国内 Ⅱ 既治療HCV 感染患者(再燃例)による 安全性・有効性・薬物動態試験 (バニプレビル+PEG2a+RBV) pPMF 製剤 100 WL00033332 WL00033773 029 海外 Ⅰ 肝生検試験 PMF( )製剤 150 WL00034194 048 海外 Ⅰ 肝生検試験 PMF( )製剤 150 WL00046346 043 国内 Ⅲ 未治療慢性HCV 感染患者による安全 性・有効性試験 (バニプレビル+PEG2b+RBV) PMF( )製剤 150 WL00036335 WL00034194 WL00046346 044 国内 Ⅲ 既治療慢性HCV 感染患者(再燃例)に よる安全性・有効性試験 (バニプレビル+PEG2b+RBV) PMF( )製剤 150 WL00036335 WL00046346 045 国内 Ⅲ 既治療慢性HCV 感染患者(無効例)に よる安全性・有効性試験 (バニプレビル+PEG2b+RBV) PMF( )製剤 150 WL00036335 WL00046346 PEG: ペグインターフェロン α、 RBV: リバビリン † 製剤中のバニプレビル含有量
付録2.7.1: 2 生物薬剤学的試験の要約 試験番号 (実施場所) 開発相 目的 (薬物動態に関連した目的) 試験デザイン 製剤/ 製剤番号 用量 登録被験者数/ 解析被験者数 (性別) 被験者タイプ 平均年齢(範囲) 結果及び考察 006 (Overseas) Phase 1
1) Assess and compare the plasma PK parameters of vaniprevir when administered as the STD FCT and FFP in healthy adult subjects.
2) Assess and compare the plasma PK parameters of vaniprevir when administered as the ARG40 FCT and FFP in healthy adult subjects Open-label, 2-part, multi-panel, randomized, multi-period, partially fixed-sequence, crossover study 100 mg FFP/ WL00025551, 100 mg STD FCT/ WL00027170, 100 mg ARG40 FCT/ WL00027207 Treatment A: 100 mg FFP, Treatment B: 100 mg STD FCT, Treatment C: 600 mg FFP, Treatment D: 600 mg STD FCT, Treatment E: 100 mg ARG40 FCT, Treatment F: 600 mg ARG40 FCT Part 1: 28/24 (23M/5F) Healthy subject 32.0 (18-45) Part 2: 12/12 (11M/1F) Healthy subject 36.4 (20-45)
1) Plasma AUC0-∞for 100 mg and 600 mg STD FCT are not
similar to respective doses of the FFP.
2) Plasma AUC0-∞for 100 mg and 600 mg ARG40 FCT are not
similar to respective doses of the FFP.
3) Administration of 100 mg STD FCT under fed conditions resulted in 59% lower AUC0-∞and 74% lower Cmaxthan when
administered under fasting conditions.
4) Administration of 600 mg vaniprevir STD FCT under fed conditions resulted in a 135% higher AUC0-∞than when
administered under fasting conditions (comparison of Cmaxwas too
imprecise to make any conclusion).
5) The administration of 100 mg vaniprevir ARG40 FCT under fed conditions resulted in a similar AUC0-∞and Cmaxcompared to when
administered under fasting conditions.
6) Administration of 600 mg vaniprevir ARG40 FCT under fed conditions resulted in a 151% higher AUC0-∞than when
administered under fasting conditions (comparison of Cmaxwas too
imprecise to make any conclusion).
7) The administration of 600 mg vaniprevir FFP under fed conditions resulted in similar AUC0-∞and 39% lower Cmax
compared to when administered under fasting conditions.
8) Single oral dose administrations of vaniprevir FCTs are safe and well tolerated in healthy subjects.
付録2.7.1: 2 生物薬剤学的試験の要約(続き) 試験番号 (実施場所) 開発相 目的 (薬物動態に関連した目的) 試験デザイン 製剤/ 製剤番号 用量 登録被験者数/ 解析被験者数 (性別) 被験者タイプ 平均年齢(範囲) 結果及び考察 014 (Overseas) Phase 1
1) Assess and compare the plasma PK parameters of vaniprevir when administered as nLFC1, nLFC2, pPMF and FFP in healthy adult subjects under fasted conditions. 2) Obtain preliminary plasma PK data following single-dose oral administration of vaniprevir in fasted and fed states for nLFC1, nLFC2 and pPMF in healthy adult subjects.
Open-label, 2-panel, randomized, 7-period, crossover (a 4-period crossover followed by a 3-period crossover), single-dose study
100 mg FFP/ WL00029579, 100 mg nLFC1/ WL00029737, 100 mg nLFC2/ WL00029738, 100 mg pPMF/ WL00029892 Treatment X: 100 mg FFP, Treatment A: 100 mg nLFC1, Treatment B: 100 mg nLFC2, Treatment C: 100 mg pPMF Treatment Y: 600 mg FFP, Treatment D: 600 mg nLFC1, Treatment E: 600 mg nLFC2, Treatment F: 600 mg pPMF Panel A: 12/12 (7M/5F) Healthy subject 36.0 (21-44) Panel B: 12/12 (8M/4F) Healthy subject 40.6 (33-45)
1) The plasma AUC0-∞values following administration of 100 mg
and 600 mg doses of the nLFC1, nLFC2 and pPMF are similar to those following administration of the same doses of the FFP under fasted conditions.
2) Vaniprevir pPMF has the most similar overall exposure relative to FFP at both the 100 mg and 600 mg doses relative to nLFC1 and nLFC2 under fasted conditions, and has been selected for further development.
3) Administration of 100 mg dose of vaniprevir with a standard high-fat breakfast resulted in similar overall exposure (AUC0-∞) for
nLFC1, nLFC2 and pPMF; similar peak exposure for nLFC1, but lower peak exposure for nLFC2 and pPMF, relative to the 100 mg in the fasted state.
4) Administration of the 600 mg dose of vaniprevir with a standard high-fat breakfast resulted in similar overall exposure (AUC0-∞) for
付録2.7.1: 2 生物薬剤学的試験の要約(続き) 試験番号 (実施場所) 開発相 目的 (薬物動態に関連した目的) 試験デザイン 製剤/ 製剤番号 用量 登録被験者数/ 解析被験者数 (性別) 被験者タイプ 平均年齢(範囲) 結果及び考察 015 (Overseas) Phase 1
1) Assess the safety and tolerability of single intravenous (IV) doses of vaniprevir.
2) Estimate the bioavailability of single oral doses of 100 mg and 600 mg of vaniprevir in healthy subjects.
3) Determine the effect of labeling vaniprevir with stable isotope (8 atoms of deuterium per molecule) on the PK of IV vaniprevir. Partially-blinded, randomized, placebo-controlled, 3-period, partially-fixed sequence crossover study HSSV 1mg/mL – 5 mL Vial/ WL00030386, HSSV 1mg/mL - 5 mL Vial/ WL00031828, HSSV Placebo - 5 mL Vial/ WL00032061, 100 mg FFP/ WL00029579 Treatment A: 10 mg IV labeled with deuterium + oral dose of 100 mg,
Treatment B: 10 mg IV labeled with deuterium + oral dose of 600 mg, Treatment C: 10 mg IV labeled with deuterium + 10 mg IV without deuterium, Treatment D:
Closely matching placebo for vaniprevir 10 mg IV + oral dose of 100 mg, Treatment E:
Closely matching placebo for vaniprevir 10 mg IV + oral dose of vaniprevir 600 mg,
Treatment F:
Closely matching placebo for 2 doses of 10 mg IV
12/12 (4M/8F) Healthy subject 57.2 (32-76)
1) Vaniprevir is generally safe and well-tolerated when administered to healthy male and female subjects by IV infusion at doses up to 20 mg.
2) The absolute bioavailability of vaniprevir is 7.9% and 33.0% following single oral dose administration of 100 and 600 mg vaniprevir respectively.
付録2.7.1: 2 生物薬剤学的試験の要約(続き) 試験番号 (実施場所) 開発相 目的 (薬物動態に関連した目的) 試験デザイン 製剤/ 製剤番号 用量 登録被験者数/ 解析被験者数 (性別) 被験者タイプ 平均年齢(範囲) 結果及び考察 021 (Japan) Phase 1
Evaluate the relative bioavailability of pPMF of vaniprevir compared to FFP in Japanese healthy adult males. Open-label, 2-part, 2-panel, 2-period, randomized crossover study 100 mg FFP/ WL00029579, 100 mg pPMF/ WL00030680 Treatment A: 300 mg FFP, Treatment B: 300 mg pPMF, Treatment C: 600 mg FFP, Treatment D: 600 mg pPMF, Treatment E: 100 mg pPMF Part 1: 24/21 (24M/0F) Healthy subject 30.2 (21-44) Part 2: 11/10 (11M/0F) Healthy subject 31.1 (22-44)
1) No statistically significant difference between FFP and pPMF was observed for AUC0-∞, Cmax, Tmaxand t1/2
2) Vaniprevir has non-linear pharmacokinetics in healthy adult Japanese males in this dose range of 100 to 600 mg.
032 (Overseas)
Phase 1
Compare the PK profile following single-dose administration of
PMF-formulation to nLFC4 and nLFC5 formulations.
Open-label, 2-part, randomized, multiple-period, crossover study (part 1: 6 period, part 2: 4 period)
150 mg PMF ( ) / WL00034194, 150 mg nLCF4/ WL00034585, 150 mg nLCF5/ WL00034584 Treatment X: 600 mg (4 x 150mg) PMF- , Treatment A: 600 mg (4 x 150mg) nLFC4, Treatment B: 600 mg (4 x 150mg) nLFC5, Treatment X (fed): 600mg (4 x 150mg) PMF- following standard high-fat breakfast, Treatment A (fed) : 600 mg (4 x 150mg) nLFC4 following standard high-fat breakfast,
Treatment B (fed):
600 mg (4 x 150mg) nLFC5 following standard high-fat breakfast Part 1: 18/18 (5M/13F) Healthy subject 38.1 (27-45) Part 2: 16/16 (5M/11F) Healthy subject 38.3 (27-45)
The AUC0-∞and Cmaxfor nLFC4 and nLFC5 formulations
are generally similar to those of PMF- formulation following a single 600 mg dose under fasted conditions Administration of 600 mg with a high-fat breakfast increases AUC by ~50% for both the PMF- and nLFC5 formulations, but has little effect on AUC of the nLFC4 formulation.
Administration of 600 mg with a high-fat breakfast increases Cmaxby ~45% for the PMF- formulation,
but has little effect on Cmaxof nLFC4 and nLFC5
formulations.
The AUC0-∞for the nLFC4 formulation is generally similar
to, but the Cmaxis ~35% lower than, that of
PMF-formulation following a single 300 mg dose under fasted conditions.
Administration of 300 mg with a high-fat breakfast increases the AUC0-∞for both nLFC4 and PMF- by
付録2.7.1: 2 生物薬剤学的試験の要約(続き) 試験番号 (実施場所) 開発相 目的 (薬物動態に関連した目的) 試験デザイン 製剤/ 製剤番号 用量 登録被験者数/ 解析被験者数 (性別) 被験者タイプ 平均年齢(範囲) 結果及び考察 049 (Japan) Phase 1
Evaluate the effects of a standard Japanese breakfast on Vaniprevir pharmacokinetics in healthy Japanese subjects.
Open-label, randomized, 2-period crossover study 150 mg PMF ( )/ WL00042510 Treatment A: 300 mg PMF , Treatment B (fed): 300mg PMF
capsule following standard
Japanese breakfast 16/14 (12M/4F)
Healthy subject 28.1 (21-35)
1) The AUC0-∞, AUC0-12 hrand Cmax following
administration of vaniprevir after meal were higher than those following administration after an overnight fast. 2) No significant changes in C12hr, and Tmax
付録2.7.1: 3 生物薬剤学的試験の薬物動態データの表 試験番号 (実施場所), 開発相 試験目的 (薬物動態) 試験デザイン 被験者 † 投与方法 (用量用法・製剤) N GM(95% CI) GMR(90% CI) AUC0-∞ (μM•hr) Cmax (μM) Tmax‡ (hr) t1/2§ (hr) AUC0-∞ Cmax 006 (Overseas) Phase 1 Part 1: To assess and compare the plasma PK parameters of vaniprevir when administered as STD FCT, or ARG40 FCT relative to that of FFP, and to obtain plasma PK data of STD FCT and ARG40 FCT in the fasted and fed states. Part 2: To obtain plasma PK of FFP in the fasted and fed states. Open-label, 2-panel, randomized, multi-period, partially fixed -sequence, crossover study Part 1: Non-Japanese healthy adult 28/24 (23M/5F) 32.0 (18-45) Part 2: Non-Japanese healthy adult 12/12 (11M/1F) 36.4 (20-45) Biocomparison between STD FCT vs. FFP Vaniprevir 1×100 mg FFP (SD, P.O., Fasted) 13 (0.190, 0.330)0.250 (0.072, 0.133)0.098 (1.00, 3.00)1.00 (26)5.12 NA NA Vaniprevir 6×100 mg FFP (SD, P.O., Fasted) 15 (2.98, 5.69)4.12 (1.07, 2.10)1.50 (1.50, 4.00)2.00 (36)3.77 NA NA Vaniprevir 1×100 mg STD FCT (SD, P.O., Fasted) 12 0.143|| (0.107, 0.193) 0.036 (0.026, 0.050) 2.00 (1.00, 4.00) 3.25|| (61) 0.57¶ (0.45, 0.74) 0.37¶ (0.26, 0.53) Vaniprevir 6×100 mg STD FCT (SD, P.O., Fasted) 14 (0.480, 0.929)0.668 (0.142, 0.284)0.200 (1.00, 4.00)2.00 (41)4.18 (0.13, 0.21)0.16¶ (0.09, 0.19)0.13¶
Biocomparison between ARG40 FCT vs. FFP Vaniprevir 1×100 mg FFP (SD, P.O., Fasted) 13 (0.181, 0.347)0.251 (0.071, 0.138)0.099 (1.00, 3.00)1.00 (26)5.12 NA NA Vaniprevir 6×100 mg FFP (SD, P.O., Fasted) 15 (3.11, 5.36)4.08 (1.09, 2.03)1.49 (1.50, 4.00)2.00 (36)3.77 NA NA Vaniprevir 1×100 mg ARG40 FCT (SD, P.O., Fasted) 12 (0.118, 0.232)0.165 (0.037, 0.072)0.052 (1.00, 4.33)3.00 (46)3.31 (0.46, 0.95)0.66†† (0.32, 0.84)0.52†† Vaniprevir 6×100 mg ARG40 FCT (SD, P.O., Fasted) 13 (1.00, 1.85)1.36# (0.297, 0.578)0.414 (1.00, 4.00)3.00 (45)4.39# (0.25, 0.45)0.33†† (0.20, 0.38)0.28††
†Type of subject, Number of subject: Entered/Analyzed(Sex), Age: Mean(Range), ‡Median(range), §Geometric mean(geometric CV%), ||N=11, ¶Geometric mean ratio(STD FCT/


![表 2.7.2: 3 非日本人健康男性に[ 14 C]バニプレビル 600 mg(約 200 μCi)を単回経口投与した際 の総放射能及びバニプレビルの薬物動態並びに放射能回収率(012 試験) 血漿中バニプレビル及び総放射能の薬物動態パラメータの比較 薬物動態 パラメータ バニプレビル 総放射能 バニプレビル/総放射能 rMSE ‡ 幾何平均 (95%信頼区間) 幾何平均 (95%信頼区間) 幾何平均比 (95%信頼区間) AUC 0-last † (μM·hr) 3.17 (2.05, 4.91) 3.](https://thumb-ap.123doks.com/thumbv2/123deta/6402278.638858/73.892.105.785.185.503/バニプレビルバニプレビルバニプレビルバニプレビルバニプレビル.webp)

![表 2.7.2: 10 被験者集団間のバニプレビルの血漿中曝露量の差の根拠として PBPK 解析で検討した日本人と非日本人間の 解剖学的、生理的因子及び酵素発現量の違い(文献情報) 比較 PBPK モデルにおける変動パラメ ータ PBPK モデルで予測されたバニプレビルの AUC の幾何平均比 統合薬物動態解析で認められた AUC の変化率 日本人対非日本人の 健康被験者: シナリオ1 ・これらの集団の PK データが得 られた臨床試験に含まれる性別、年齢、身長及び体重の分布 ・日本人[資料5.4: 39]](https://thumb-ap.123doks.com/thumbv2/123deta/6402278.638858/97.892.106.788.204.579/バニプレビルモデルバニプレビル日本人シナリオデータ含まれる.webp)