タンパク質のリガンド結合部位予測システムの開発
6
0
0
全文
(2) Vol.2012-BIO-28 No.19 2012/3/29. 情報処理学会研究報告 IPSJ SIG Technical Report. ギーの最小化は sander を用いて 10000 ステップ実行した. 続いて,AMBER に含まれる sander を用いて系の圧力を一定のまま,温度を 0K から. 300K まで 30ps かけて上昇させる.その後 20ps の等温の MD シミュレーションを行う.こ れは,水分子を系の中で緩和させつつ,一般的な温度にするために実行する.. 2.2 水分子のダイナミクスの解析 2.1 の過程を経たタンパク質と水分子の系について,評価のための MD シミュレーション を行う.MD は温度が一定となる NPT アンサンブルを採用する.1 ステップの時間幅は,. 2fs とした. MD シミュレーションの結果から 10ps 毎に,タンパク質と水分子の各原子の座標を PDB ファイルとして出力する. 水分子の評価の方法は次のようにした.タンパク質の周辺に 3 次元のグリッド上を配置 する.タンパク質と水からなる空間に 0.9˚ A 間隔のグリッドを作る.各グリッドを中心とす ˚ る一辺 3.6A の立方体を考え,その立方体内に水分子を構成する酸素原子が存在するかどう かをカウントする.各グリッドを時刻毎に調査して,全シミュレーション時間に対する水分. 図 1 システムのフローチャート Fig. 1 Prediction System flowchart.. 2.1 準. 子の存在時間の割合を求める.. 備. 2.3 タンパク質表面の幾何学的なポケット探索. 分子動力学法(MD : molecular dynamics)シミュレーションを用いることで,対象とな. タンパク質表面の幾何学的手法を用いたポケット探索を行なう.本研究では,プローブの 半径は 1.6˚ A,グリッドの大きさは 0.9˚ A として Ligsite アルゴリズムを適用した.. るタンパク質の周囲の水分子のダイナミクスを獲得する. そのための準備として,以下のような手順で解析に使うタンパク質データを作成した.. Ligsite は,タンパク質の形状に基づいてポケット探索を行なうアルゴリズムである.タ. Protein Data Bank (PDB)11) 形式のファイルから,リガンドや水など標的のタンパク質に. ンパク質の周囲にグリッドを配置して,グリッドを走査していき,ポケットを構成するグ. 含まれない原子を除去する.. リッドとグリッドの集合を判定する.手順は次の通りである.. AmberTools8) に含まれるソフトウェア LEaP を用いて次のような手続きを行なった.. (1). タンパク質の周りに 3 次元のグリッドを配置する.. (1). タンパク質の PDB ファイルを読み込む. (2). x 軸方向に沿ってスキャンを行なう.. (2). タンパク質に水素原子を付加. (3). この時に,タンパク質-空間-タンパク質となるグリッドをチェックする. ナトリウムイオン Na+,もしくは塩化物イオン Cl-を配置し,全体の電荷の総和を 0. (4). (3). にする. (4). 水分子をタンパク質の外側へ直方体状に配置する.. (5). トポロジーファイルと原子の座標ファイルを出力を行う. y 軸方向,z 軸方向,および立方体の対角線方向(4 方向)についても同様にスキャ ンを行ない,タンパク質-空間-タンパク質となるグリッドをチェックする.. (5). 7 方向のスキャンを行なった後に,5 回以上チェックされたグリッドをポケットを構 成するグリッドとする.. 次に,系の原子位置を最適化しひずみを取るためのエネルギー最小化を行なう.エネル. (6). 2. 最後に隣接したグリッドを同じクラスタとみなし、ポケットの大きいもの、つまりグ. c 2012 Information Processing Society of Japan ⃝.
(3) Vol.2012-BIO-28 No.19 2012/3/29. 情報処理学会研究報告 IPSJ SIG Technical Report Protein. Ligand(log P). Protein. Ligand(log P). 3FR2 1AJX 2JK7 1JK3 1RM8 1FKF 2VN1 1UY6 3S92 1T3R. 8CA(4.22) AH1(4.05) BI6(3.62) BAT(3.23) BAT(3.23) FK5(3.19) FK5(3.19) PU3(2.96) JQ1(1.94) 017(1.89). 3T1M 2LIW 3BIB 3N2U 2QEO 1CEB 1OW4 2HL0 1HPK 3CYH. DQT( 1.79) MAH(-0.88) PSF(-1.02) D3X(-1.2 ) LNR(-1.4 ) AMH(-1.42) GOL(-1.93) A3S(-1.93) ACA(-2.69) PRO(-2.71). 表 1 PDB-ID と結合するリガンド Table 1 PDB-ID and binding ligand. リッド数が多いクラスタの上位 10 個を出力する。. 2.4 結果の統合 図 2 Ligsite と提案手法のポケット領域のグリッドの割合の比較 Fig. 2 The graph shows the comparison of percentage of the grid in estimated pocket between only Ligsite and proporsal method.number of low information entropy area of water around protein.. MD シミュレーションから得た水分子の情報と,ポケット探索の結果を統合する.水分子 のダイナミクスの情報は,グリッドを基準とした解析を行なう事で,ポケット探索と統合す る事が容易になる.Ligsite によってポケットと判定され,各グリッドにおける水分子の存. BER118) の sander.MPI を用いた.長距離の静電相互作用には Particle Mesh Ewald(PME) ˚ に設定した. 法を用いて,カットオフ距離は 10A. 在時間が全体時間に対する割合の閾値を越えたグリッドの集合をリガンド結合部位の候補と する.. 解析用の分子動力学シミュレーションは,本学のスーパーコンピュータ TSUBAME2.0 を. 3. 評 価 実 験. 用いて 32∼128 並列にて sander.MPI を実行した.水分子のみの動きを観察するためタン. 3.1 実験の条件. パク質を固定し,10ns 分の時系列データを作成した.観測時間の閾値は全体の 60%とした.. 本研究では,対象となるタンパク質として PDB から 20 種類 (1AJX, 1CEB, 1FKF,. 3.2 結. 果. 1HPK, 1JK3, 1OW4, 1RM8, 1T3R, 1UY6, 2HL0, 2JK7, 2LIW, 2QEO, 2VN1, 3BIB,. タンパク質毎のグリッド数削減の割合を示したのが,図 2 である.2.1 の過程が終わった. 3CYH, 3FR2, 3N2U, 3S92, 3T1M) を準備した.これらのタンパク質は,リガンドの疎水. 状態のタンパク質に対し Ligsite アルゴリズムのみを適用して得た結合部位候補のグリッド. 性・親水性による違いを調べるため,分配係数 (log P) の値を基準に選択した.分配係数と. の数を 1 として,提案手法で得られた候補のグリッドとの比較を行なった.1FKF を除い. は,物質を油性の溶媒と水に溶かした時の,濃度の比の対数をとったものである.これが負. た全てのタンパク質で,4 割あるいはそれ以上の削減が見られた.割合で見て,もっとも多. 値ならば親水性であり,正値ならば疎水性となる.分配係数は,分子の構造から予測する事. く削減したのは 3T1M で,Ligsite のみの場合の 14%まで,グリッド数を削減した.. が可能であり,今回は ALOGPS12) の値を参考にした.各タンパク質に結合するリガンド. 次に予測したポケットの数を 表 2 に示す.Ligsite では 10 個のポケットが予測されるが,. を表 1 に示す.結合するリガンドが疎水性のタンパク質が 11 種,親水性のリガンドが 9 種. 提案手法によって,平均して 5.3 個まで絞りこむ事ができた.1FKF はグリッドの削減数が. である.. 少なかった事もあり,ポケットの数も削減する事ができなかった.同様に,候補のグリッド. 周囲に配置する水分子は TIP3P モデルを用いた.分子動力学シミュレーションには,AM-. を最も多く削減した 3T1M は,予測ポケットの個数も 2 個と大きく削減された.. 3. c 2012 Information Processing Society of Japan ⃝.
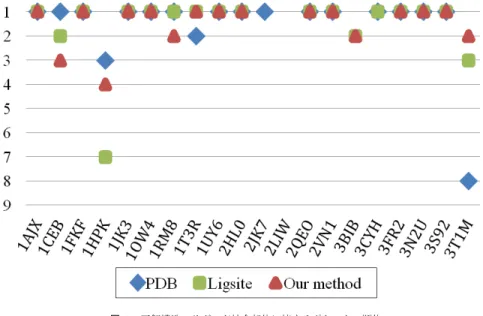
(4) Vol.2012-BIO-28 No.19 2012/3/29. 情報処理学会研究報告 IPSJ SIG Technical Report Protein 3FR2 1AJX 2JK7 1JK3 1RM8 1FKF 2VN1 1UY6 3S92 1T3R. Pocket 4 6 6 6 6 9 6 6 3 5. Protein 3T1M 2LIW 3BIB 3N2U 2QEO 1CEB 1OW4 2HL0 1HPK 3CYH ave.. Pocket 2 5 3 5 7 8 4 5 5 6 5.3. 表 2 予測したポケットの数 Table 2 The number of prediction pocket. 図 3 は,リガンド結合部位を正しく予測できたかを見るため,予測結果をグリッドの数 を基準とした順位をつけ,第何位のポケットが結合部位の領域に現れるかしめしたものであ る.これは,PDB の web サイトからダウンロードしたデータに直接 Ligsite を適用した結 果(PDB),2.1 の過程が終わった状態のタンパク質に Ligsite を適用した結果(Ligsite),. 図 3 正解構造のリガンド結合部位に接するポケットの順位 Fig. 3 The graph shows rank of the estimated pocket in contact with the correct binding site. Red markers show proposal, green show Ligsite only and blue show PDB download data.. 水分子の観測頻度を取り入れた手法(提案手法)の 3 種類で比較を行なった.. PDB に直接適用した場合が最も良いタンパク質は 3 種類,Ligsite のみ適用した結果が 最も良いタンパク質は 2 種類,提案手法による改善が見られたのは 1 種類であった.13 種 類のタンパク質では,Ligsite と提案手法の順位が変わらなかった.1HPK では Ligsite の. 端に狭く、水分子が十分に入ることが出来なかったことが原因であると考えられる.また,. み適用の場合に,PDB の結果より大きく順位を落としているが,提案手法によって順位が. 結合部位周辺の残基の多くは親水性であったが,疎水性の残基も存在していた.これによっ. 改善された.. て,水分子が留まる事が難しくなった事が考えられる.. 最も良い改善となったのは 3T1M である.PDB のデータに直接適用した場合は,正解の. 分配係数による違いについては,ポケット数の削減や順位の変動の結果から,これによる. 領域は第 8 位であった.準備の過程が終わった状態に Ligsite を適用した場合に第 3 位まで. 差異はないと考えられる.. 上昇し,提案手法では第 2 位となった.3T1M では,Ligsite による予測の多くが,タンパ. 提案手法と Ligsite を比較すると,提案手法によって,順位が下がったタンパク質が 2 種. ク質の折りたたまれた内部に現れている (図 4).そのため,その領域には水分子が入り込む. 類 (1CEB, 1RM8) 存在した.しかしどちらも下がった順位は一つであり,問題ないと考え. 事ができず,リガンド結合部位となる領域の順位を上げた.. られる.これは,本システムを結合部位の候補を提案するシステムと捉えるならば,多少の. 提案手法では2つのタンパク質において,予測がうまくいかなかったがこの原因について. 順位の変動より,提案する結合領域の個数を削減する事,および,削減後の結果に正解の領. 考える.2LIW は全ての手法で正解となる領域の予測ができなかった.これは結合部位の形. 域が含まれている事が重要だと考えられるからである.. 状が平らに近い事が原因だと考えられる (図 5).3CYH では,準備の過程を終えたタンパ ク質に Ligsite のみを用いた手法が正解の領域を予測できた.立体構造では、結合領域が極. 4. c 2012 Information Processing Society of Japan ⃝.
(5) Vol.2012-BIO-28 No.19 2012/3/29. 情報処理学会研究報告 IPSJ SIG Technical Report. 考えられる.またリガンドの親水性・疎水性による結果の差異は見られなかったため,多く のタンパク質について本システムを適用する事が可能であると示された. 二つの手法を組み合わせる事によって,欠点を補い合う事が可能となり,リガンド結合部 位の予測の結果向上につながったと考えられる.. 参. 考. 文. 献. 1) Oprea, T.I., Davis, A.M., Teague, S.J. and Leeson, P.D.: Is There a Difference between Leads and Drugs? A Historical Perspective, J. Chem. Inf. Comput. Sci., Vol.41, No.5, pp.1308–1315 (2001). 2) Shuker, S.B., Hajduk, P.J., Meadows, R.P. and Fesik, S.W.: Discovering HighAffinity Ligands for Proteins: SAR by NMR, Science 29 November 1996, Vol.274 No.5292 pp.1531–1534 (1996). 3) Kuntz, I.D.: Structure-Based Strategies for Drug Design and Discovery Science 21 August 1992, Vol.257 No.5073 pp.1078–1082 (1992). 4) Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L., Clardy, J.: Atomic Structure of FKBP-FK506, an Immunophilin-Immunosuppressant Complex Science 10 May 1991, Vol.252 No.5007 pp.839–842 (1991). 5) Abel, R., Young, T., Farid, R., Berne, B.J. and Fresner, R.A.: Role of the ActiveSite Solvent in the Thermodynamics of Factor Xa Ligand Binding J. Am. Chem. Soc., Vol.130, No.9, pp.2817–2831 (2008). 6) Hendlich M, Rippmann F, Barnickel G.: LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol. Graph. Model. Dec;15(6):359-63, 389 (1997). 7) Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E. III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules.Comp. Phys. Commun., (1995) 91, 1―41. 8) D.A. Case, T.A. Darden, T.E. Cheatham, III, C.L. Simmerling, J. Wang, R.E. Duke, R. Luo, R.C. Walker, W. Zhang, K.M. Merz, B.P. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvry, K.F. Wong, F. Paesani, J. Vanicek, J. Liu, X. Wu, S.R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D.R. Roe, D.H. Mathews, M.G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, and P.A. Kollman (2010), AMBER 11, University of California, San Francisco. 9) Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L., Clardy, J.: Atomic Structure of FKBP-FK506, an Immunophilin- Immunosuppressant Com-. 図 4 3T1M の結果.タンパク質をリボン形式で示して いる.青点が Ligsite のみの結果,オレンジが提 図 5 2LIW の複合体構造.リガンドがタンパク質上 部の平らな面に結合している. 案手法の結果. Fig. 4 The result of 3T1M is shown. blue dotFig. 5 2LIW structure is shown. ligand binding site (this figure upper) seems flat. cluster is the result of Ligsite, and orange is proposal method.. 4. ま と め 本研究では,タンパク質のリガンド結合部位を予測する方法について,水分子のふるまい とポケット探索を用いた手法で,実験・評価を行なった.その結果,候補となるポケットの 数を Ligsite のみの手法と比較して,平均で約半数に削減する事ができた.これは,ポケッ ト探索のみの場合では,熱揺らぎによる偶然できたくぼみを水分子のダイナミクスを取り入 れる事で,候補から外す事ができるからと考えられる.一方で,水分子のダイナミクスだけ では,タンパク質表面の物理化学的な性質に影響されて,凸面や平面においても,水分子 が滞在し続ける事がある.そのような場合については,ポケット探索で形状を考慮する事に よって,外す事ができる. グリッドの数で順位付けを行なったところ,提案手法と Ligsite では,ほぼ同じ順位で あった.結合部位の候補を提供するシステムとするならば,多少の順位の入替は問題ないと. 5. c 2012 Information Processing Society of Japan ⃝.
(6) Vol.2012-BIO-28 No.19 2012/3/29. 情報処理学会研究報告 IPSJ SIG Technical Report. plex, Science 10 May 1991, Vol. 252 No. 5007 pp. 839―842 (1991). 10) Abel, R., Young, T., Farid, R., Berne, B. J. and Fresner, R. A.: Role of the ActiveSite Solvent in the Thermodynamics of Factor Xa Ligand Binding J. Am. Chem. Soc., Vol. 130, No. 9, pp. 2817―2831 (2008). 11) RCSB PDB : http://www.pdb.org/pdb/home/home.do 12) Tetko, I. V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V. A.; Radchenko, E. V.; Zefirov, N. S.; Makarenko, A. S.; Tanchuk, V. Y.; Prokopenko, V. V. Virtual computational chemistry laboratory - design and description,J. Comput. Aid. Mol. Des., 2005, 19, 453-63, 13) 佐々木孝章, 関嶋政和: タンパク質中の水分子の情報エントロピーによるリガンド結合 部位予測, 第 11 回日本蛋白質科学会年会, pp. 130-130, (2011). 14) 佐々木孝章, 関嶋政和: 情報エントロピーによるタンパク質のリガンド結合部位予測, 情報処理学会第 73 回全国大会, (2011).. 6. c 2012 Information Processing Society of Japan ⃝.
(7)
図



関連したドキュメント
Likewise we show that any decomposition of the complete graph into strongly regular graphs of (negative) Latin square type is an amorphic association scheme.. We study strongly
This paper develops a recursion formula for the conditional moments of the area under the absolute value of Brownian bridge given the local time at 0.. The method of power series
Related to this, we examine the modular theory for positive projections from a von Neumann algebra onto a Jordan image of another von Neumann alge- bra, and use such projections
Answering a question of de la Harpe and Bridson in the Kourovka Notebook, we build the explicit embeddings of the additive group of rational numbers Q in a finitely generated group
modular proof of soundness using U-simulations.. & RIMS, Kyoto U.). Equivalence
Splitting homotopies : Another View of the Lyubeznik Resolution There are systematic ways to find smaller resolutions of a given resolution which are actually subresolutions.. This is
Then it follows immediately from a suitable version of “Hensel’s Lemma” [cf., e.g., the argument of [4], Lemma 2.1] that S may be obtained, as the notation suggests, as the m A
In our previous paper [Ban1], we explicitly calculated the p-adic polylogarithm sheaf on the projective line minus three points, and calculated its specializa- tions to the d-th
