九州大学学術情報リポジトリ
Kyushu University Institutional Repository
Development of engineered biomolecules
exploiting host biological systems for cancer therapy
佐々木, 光一
http://hdl.handle.net/2324/4060130
出版情報:九州大学, 2019, 博士(工学), 課程博士 バージョン:
Ph.D. Thesis
Development of engineered biomolecules exploiting host biological systems
for cancer therapy
March 2020
Department of Materials Physics and Chemistry Graduate School of Engineering
Kyushu University
Koichi Sasaki
Table of Contents
Abstract ... 1
Chapter 1; General Introduction ... 3
1.1 Targeted drug delivery for cancer therapy ... 3
1.1.1 Active targeting ... 4
1.1.2 Passive targeting ... 6
1.1.3 Acquired resistance to targeted drug delivery ... 7
1.2 Host immune system and anti-cancer therapeutic agents ... 9
1.2.1 Therapeutics for immune activation ... 9
1.2.1.1 Therapeutic antibodies ... 9
1.2.1.2 Immune checkpoint inhibitor ... 12
1.2.1.3 Immune-stimulating anticancer small molecules ... 14
1.2.2 Adverse immune responses upon cancer immunotherapy ... 16
1.2.2.1 Autoimmune responses ... 16
1.2.2.2 Production of anti-drug antibody ... 17
1.3 Overview of this thesis ... 19
1.4 References ... 21
Chapter 2; Collagen-binding serum albumin as a drug conjugate carrier for treating solid tumors ... 27
2.1 Introduction ... 27
2.2 Results ... 32
2.2.2 Dox release under acidic pH conditions ... 34
2.2.3 Dox conjugates are taken up by cancer cells and retain cytotoxicity ... 35
2.2.4 Dox-CBD-SA shows similar blood plasma pharmacokinetics as aldoxorubicin and efficiently accumulates in tumors ... 37
2.2.5 Dox-CBD-SA demonstrates enhanced antitumor efficacy in the MMTV-PyMT murine breast cancer model ... 39
2.2.6 Dox-CBD-SA facilitates tumor infiltration of lymphocytes ... 40
2.2.7 Dox-CBD-SA shows reduced toxicity ... 42
2.2.8 Dox-CBD-SA in combination with anti−PD-1 antibody eradicates MC38 tumor ... 44
2.3 Discussion ... 46
2.4 Summary ... 49
2.5 Experimental procedures ... 50
2.6. Supplementary Information ... 58
2.7 References ... 66
Chapter 3; Fc-binding antibody-recruiting molecules: exploiting the majority of endogenous IgG for anti-cancer immune responses ... 72
3.1 Introduction ... 72
3.2 Results ... 80
3.2.1 Fc-ARM2 shows stronger binding to IgG-Fc and improved
3.2.2 Human IgG1 redirected to tumor cells by Fc-ARM activates
NK cells without antigen–Fab interactions ... 82
3.2.3 Fc-affinity of Fc-ARM positively correlates with NK cell activation by the recruited antibody ... 84
3.2.4 Fc-ARM interacts with IVIG to enhance blood retention time and tumor accumulation in vivo ... 85
3.2.5 Fc-ARM2 suppresses tumor growth in combination with IVIG ... 87
3.3 Discussion ... 89
3.4 Summary ... 90
3.5 Experimental procedures ... 91
3.6. Supplementary Information ... 97
3.7 References ... 108
Chapter 4; Fc-ARM as a potential inhibitor of off-target effects of ADCC ... 114
4.1 Introduction ... 114
4.2 Results and Discussion ... 117
4.2.1 Fc-ARM enhances the accumulation of anti-EGFR on EGFR/FR-a double positive cells ... 117
4.2.2 Fc-ARM inhibits ADCC activity of anti-EGFR against
EGFR/FR-a double positive cells ... 120
4.2.3 Multiple markers confirm the inhibitory activity of Fc-ARM
4.3 Summary ... 125
4.4 Experimental Procedures ... 126
4.5 Supplementary Information ... 130
4.6 References ... 134
Chapter 5; General Conclusions ... 136
Achievements ... 138
List of publications ... 138
List of supplementary publications ... 138
List of other publications ... 138
List of presentations ... 139
Honors/Awards ... 141
Acknowledgements ... 142
Abstract
Two ultimate goals in cancer therapy are to make cancer curable, and possibly to minimize adverse events associated with the treatments. Drug delivery system (DDS) and cancer immunotherapy are important strategies which have potential to achieve these goals. DDS in the field of cancer therapy means to deliver toxic anti-tumor agents selectively to cancer tissues, thereby enhancing therapeutic efficacy and reducing collateral damages to normal cells. DDS showed remarkable successes in the preclinical stage, however, its effectiveness has not been proven in the clinic. It is considered that cancer heterogeneity in solid tumors, as well as acquired resistance to anti-cancer agents and DDS methodologies are main obstacles mitigating the efficacy of targeted drug delivery.
Cancer immunotherapy is gaining huge attention these days. Many approaches classified into cancer immunotherapy can activate immune systems to multiple tumor antigens, showing huge potential to overcome cancer heterogeneity and acquired drug resistance. Indeed, there are multiple patients diagnosed refractory to conventional therapies but have been cured after cancer immunotherapeutic treatments. On the other hand, there are emerging problems needed to be addressed for better clinical successes of cancer immunotherapy. For example, some cancer immunotherapeutics are reportedly not effective in patients with less tumor-infiltrating lymphocytes. Treatment-associated immune responses to normal cells are life-threatening side effects of cancer immunotherapy. In addition, antibody production against macromolecular therapeutics causes reduced therapeutic efficacy and additional side effects. In this thesis, I sought to address these issues through development of a novel protein for cancer drug delivery, and development of less immunogenic cancer immunotherapeutics with antibody’s effector functions.
In chapter 2, we designed a fusion protein of serum albumin (SA) and the collagen binding domain (CBD) of von Willebrand factor, namely CBD-SA. CBD-SA passively targets tumor tissues through enhanced permeability and retention (EPR) effect, and actively target collagens within tumor microenvironment. Doxorubicin (Dox, a classic anti-cancer agent) conjugated CBD-SA
into tumor, and synergistically eradicated a murine colon carcinoma model in combination with anti-programmed cell death-1 (PD-1) checkpoint inhibitor antibody.
In chapter 3, we intended to develop a class of synthetic alternatives of therapeutic antibodies.
Specifically, bispecific conjugates of a cyclic peptide which specifically binds to IgG-Fc and a folic acid as a tumor targeting ligand were synthesized. We named them “Fc-binding antibody- recruiting molecules (Fc-ARMs)”. Fc-ARM is expected to target more than 80% of endogenous IgG in the circulation and redirect them to caner tissues. Fc-ARM successfully redirected IgG onto folate receptor-a (FR-a) positive cancer cells, and showed anti-tumor efficacy both in vitro and in vivo.
In chapter 4, we investigated a potential of Fc-ARM as a specific inhibitor of antibody-dependent cell-mediated cytotoxicity (ADCC). Combinatorial use of anti-epidermal growth factor receptor (EGFR) antibody and Fc-ARM against EGFR+ FR-a- cells showed comparable ADCC efficacy with anti-EGFR alone, whereas showed reduced ADCC efficacy against EGFR+ FR-a+ cells.
These results suggest that Fc-ARM could be potentially used to reduce side effects of therapeutic antibodies against normal cells expressing a defined antigen.
The critical information provided in this thesis would give insights into DDS for overcoming acquired drug resistance as well as adverse immune responses associated with cancer immunotherapy.
Chapter 1; General Introduction
1.1 Targeted drug delivery for cancer therapy
Cancer is one of the major causes of death in developed countries. About 30% of adults develop invasive cancer from birth to death in the United States 1. In Japan, about a half of adults develop cancer during their lifetime 2. Chemotherapy represents major methodologies for treating cancers, which enables to fight with multiple malignant tissues disseminated in entire body. Classic chemotherapeutic agents diffuse systemically in vivo and inhibit cell division. Because tumor cells replicate relatively faster than normal cells, anti-cancer agents preferentially attack cancer cells.
Well known adverse events during chemotherapy are myelosuppression, vomiting, and hair-loss, all of these are attributed from damages to relatively rapid growing cells in bone marrow, gastrointestinal tract, and hair follicles. In theory, it is preferable to administer anti-cancer agents to patients as much as possible to minimize the risk of acquired drug resistance and relapse of cancer. However, actual amount of drug dosage is limited to maximum tolerated dose (MTD), which refers to the highest dose of a treatment without unacceptable toxicity. Therefore, development of a methodology called targeted drug delivery, which is intended to locally enhance the concentration of administered drugs where they are needed in human body, is of exceptional importance for improved therapeutic outcome of cancer. Currently, methods for achieving targeted drug delivery are classified into two types; active targeting and passive targeting.
1.1.1 Active targeting
Actively targeted drugs possess at least one moiety which binds to specific proteins, lipids, or sugars. Affinity ligands enhance bio-distribution of drugs to the targets while minimizing interaction between the drugs and off-target molecules, thereby contributing to improved therapeutic efficacy and reduced side effects. Generally speaking, molecules overexpressed, or specifically expressed in cancer tissues are targeted for treating cancer. Trastuzumab emtansine (T-DM1) is, for instance, a chemical conjugate of anti-human epidermal growth factor receptor 2 (HER2) antibody and the cytotoxic agent DM1 (Fig. 1.1). Anti-HER2 antibody serves as a targeting moiety and efficiently deliver DM1 to HER2 positive breast cancer tissues, showing improved therapeutic outcome in the clinic 3. In other cases, the surface of liposome bearing chemotherapeutics can be modified with affinity ligands to target a variety of cancers. Multiple types of liposomes engineered in this manner have been tested in clinical trials 4. Recent advancements in drug development enable to discover myriad of affinity ligands [such as antibodies 5-7, nanobodies (developed from camelid antibodies)8-9, affibodies (artificial proteins based on the immunoglobulin-binding domain of staphylococcal protein A)10-11, nucleic acid aptamers 12-14, peptides 15-18, and sugars 19-20] for targeting virtually any types of molecules.
Fig. 1.1. Structure of T-DM1 and mechanisms of action. After T-DM1 binds HER2, the HER2/T-DM1 complex undergoes internalization, followed by lysosomal degradation. This process results in the intracellular release of DM1 containing catabolites that bind to tubulin and prevent microtubule polymerization as well as suppress microtubule dynamic instability. T-DM1 has also been shown to retain mechanisms of action of trastuzumab, including disruption of the HER3/PI3K/AKT signaling pathway and Fcg receptor–mediated engagement of immune effector cells, which leads to antibody-dependent cellular cytotoxicity. Reprinted with the permission from ref 21.
1.1.2 Passive targeting
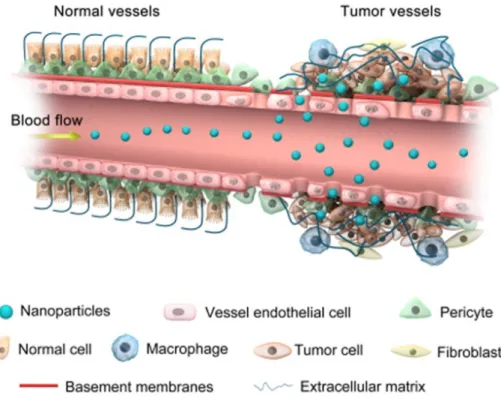
Passive targeting of tumor tissues is based on an effect called enhanced permeability and retention (EPR) effect (Fig. 1.2), first defined by Maeda et al in 1986 22. Tumor vasculature is physiologically hyperpermeable, which enables large particles to enter tumor sites. Further, lymphatic drainage is often dysfunctional in tumor tissues, allowing nanoparticles which can avoid renal clearance (larger than 40 kDa) to accumulate in tumor tissues 23. These days, most of nanoparticles (e.g. liposomes, micelles) for cancer drug delivery are fine-tuned to show EPR effect.
Other popular drug carriers (such as antibody 24, serum albumin 22, gold nanorod 25, etc.) are also thought to exhibit EPR effect.
Fig. 1.2. Accumulation of nanoparticles in tumor tissues via the EPR effect. Reprinted with the permission from ref 26.
1.1.3 Acquired resistance to targeted drug delivery
Extensive efforts by researchers have realized targeted drug delivery to solid tumors in preclinical stages. However, effectiveness of the strategy is not well proven in the clinic. For example, passive targeting of doxorubicin (Dox) through liposomal formulation did not show improved therapeutic efficacy compared with the free Dox as a first-line treatment of metastatic breast cancer 27. Cancer cells can be unresponsive to the targeted chemotherapies due to their heterogeneity and acquired resistance to both active targeting and passive targeting.
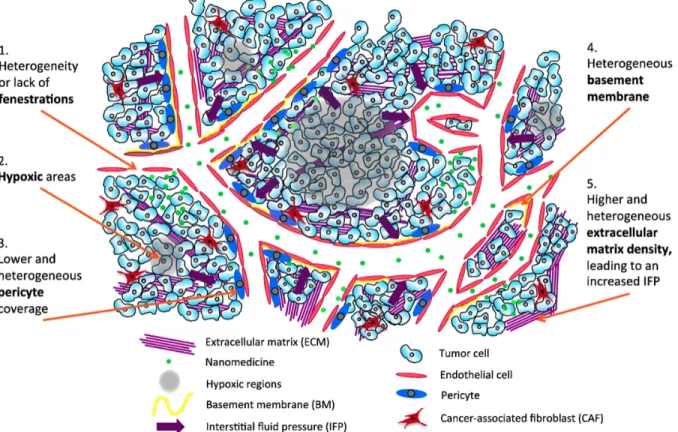
Proteins exploited for active targeting can be mutated during treatment, thereby preventing nanodrugs to bind to the targets. For example, some population of colorectal cancer patients develop substitution of serine 492 within the ectodomain of the epidermal growth factor receptor (EGFR) to arginine (Fig. 1.3), resulting in acquired resistance to anti-EGFR monoclonal antibody (cetuximab)28. Molecular and genetic heterogeneity in solid tumors is also confronting the active targeting strategy. Antigen-selective killing of cells by the active targeting strategy could work as a kind of selection processes, eventually lead growth of survivor cells which do not express target proteins29. In the case of passive targeting, its efficacy is not proven in the clinic due to heterogeneous properties of cancers. As summarized in Fig. 1.4, lots of factors such as a degree of fenestrations, hypoxic areas, pericyte coverage of vasculature, basement membrane, and the densities of extracellular matrix in tumor microenvironment are all considered to influence on the EPR effect 30. In summary, there is substantial room for improvement both in active and passive targeting to circumvent resistance to chemotherapeutic intervention.
Fig. 1.3. Cetuximab-resistant cells harbor a missense mutation (S492R) within the extracellular domain of EGFR. (a) Nucleotide sequence of the EGFR gene in DiFi cells and DiFi-derived cetuximab-resistant clones (DCR). A heterozygous mutation resulting in the substitution of a serine with an arginine at the position corresponding to amino acid 492 in the extracellular domain of EGFR is observed in DCR clones (arrows). M, mutation. (b) Structural modeling of the interaction between EGFR domain III and cetuximab, confirming the position of EGFR Ser492 at the interface. EGF is shown in purple. Reprinted with the permission from ref 28.
Fig. 1.4. Schematic representation of the passive targeting of nanomedicine (EPR effect) in
1.2 Host immune system and anti-cancer therapeutic agents
As highlighted in the 2018 Nobel prize in physiology or medicine jointly awarded to Prof. James P. Allison and Prof. Tasuku Honjo (for their discovery of cancer therapy by inhibition of negative immune regulation 31), utilization of host immune system is showing remarkable success for treating cancer in the clinic. Given that immune system is originally designed to eliminate diverse pathogens, abnormal cells, etc., immunotherapy would be an effective approach to fight with cancer heterogeneity. Here, I summarize major modalities of cancer immunotherapy and their mechanisms of actions. In addition, I will introduce the mechanisms how such therapeutics suffer from the host immune system as an emerging problem in this field.
1.2.1 Therapeutics for immune activation 1.2.1.1 Therapeutic antibodies
As a cancer immunotherapeutic, antibody drugs are extremely useful for killing tumor cells through multiple mechanisms (Fig. 1.5)32. In contrast to direct killing of tumor cells (or pro-tumor cells residing in tumor microenvironment through apoptotic signaling induction, blockade of proliferating signaling, and delivery of toxic agents), induction/enhancement of immune responses against cancer cells are the unique mechanisms of actions that are difficult to be induced by classic anti-cancer small molecules. Notably, antibody-dependent immune responses against a specific antigen could spread adaptive immune responses to other antigens33, suggesting the potential of therapeutic monoclonal antibodies (mAbs) to fight with cancer heterogeneity. As of May 2016, 23 therapeutic mAbs are approved for treatment of cancers by U.S. Food and Drug Administration (FDA) (Table 1.1). 34
Fig. 1.5. Mechanisms of tumour cell killing by antibodies. A | Direct tumour cell killing can be elicited by receptor agonist activity, such as an antibody binding to a tumour cell surface receptor and activating it, leading to apoptosis (represented by the mitochondrion). It can also be mediated by receptor antagonist activity, such as an antibody binding to a cell surface receptor and blocking dimerization, kinase activation and downstream signaling, leading to reduced proliferation and apoptosis. An antibody binding to an enzyme can lead to neutralization, signaling abrogation and cell death, and conjugated antibodies can be used to deliver a payload (such as a drug, toxin, small interfering RNA or radioisotope) to a tumour cell. B | Immune-mediated tumour cell killing can be carried out by the induction of phagocytosis; complement activation; antibody-dependent cellular cytotoxicity (ADCC); genetically modified T cells being targeted to the tumour by single- chain variable fragment (scFv); T cells being activated by antibody-mediated cross-presentation of antigen to dendritic cells; and inhibition of T cell inhibitory receptors, such as cytotoxic T lymphocyte-associated antigen 4 (CTLA4). C | Vascular and stromal cell ablation can be induced by vasculature receptor antagonism or ligand trapping (not shown); stromal cell inhibition;
attack complex; MHC, major histocompatibility complex; NK, natural killer. Reprinted with the permission from ref 32.
Drug Name Target Indication
Rituxan CD20 B-NHL, B-CLL
Herceptin EGF Breast cancer
Mylotarg CD33 AML
Campath CD52 B-CLL
Zevalin CD20 B-NHL
Erbitux EGFR Colorectal cancer
Avastin VEGF Colon cancer
Vectibix EGFR Colorectal cancer
Arzerra CD20 B-CLL
Yervoy CTLA-4 Melanoma etc
Adcetris CD30 HL, ALCL
Perjeta HER2 Breast cancer
Kadcyla HER2 Breast cancer
Gazyva CD20 B-CLL, FL
Cyramza VEGFR2 Gastric cancer
Ketruda PD-1 Melanoma, lung cancer, etc Bexxar CD19 + CD3 NHL
Opdivo PD-1 Melanoma, lung cancer, etc
Unituxin GD2 Neuroblastoma
Darzalex CD38 MM
Portrazza EGFR Lung cancer
Empliciti SLAMF7 MM
Tecentiq PD-L1 Urothelial cancer, lung cancer, etc
lymphocytic leukemia; HL = Hodgkin lymphoma; ALCL = anaplastic large cell lymphoma; FL = follicular lymphoma; MM = multiple myeloma. Modified from ref34.
1.2.1.2 Immune checkpoint inhibitor
Immune checkpoint proteins are a group of regulators which play crucial roles in self-tolerance.
As one of immune checkpoint protein, programmed cell death-1 (PD-1) was first discovered by Honjo et al in 1992 35. PD-1 is a transmembrane protein, and its expression is transcriptionally induced in activated T cells, B cells, etc 35-37. PD-L1 (a ligand of PD-1) is expressed on antigen- presenting cells (APCs) as well as nonlymphoid cells (e.g. cells in heart and lung). PD-1−PD-L interaction plays two roles to maintain peripheral tolerance38. PD-Ls on APCs induce peripheral tolerance through inhibition of the activation of T cells. PD-Ls on normal cells directly prevents effector function of T cells and contribute to maintain peripheral tolerance.
Cytotoxic T lymphocyte antigen 4 (CTLA-4) was first discovered by Golstein et al in 1987 39. CTLA-4 is also a transmembrane protein, but its expression is limited to T cells. CTLA-4 and CD28 (T cell co-stimulatory protein) share the same ligand called B7 (CD80/CD86), which is expressed on APCs. Once T cell activation is initiated by TCR signaling and co-stimulation by CD28−B7 interaction, CTLA-4 expression is upregulated and competitively inhibits CD28−B7 interaction. Furthermore, CTLA-4−B7 interaction transmits inhibitory signal to T cells40.
In addition to maintenance of self-tolerance, immune checkpoint proteins contribute to prevent overactivation of immune system. However, this system is exploited by cancer cells for immune evasion. For instance, a variety of cancer cells express PD-L1 to exploit the mechanism of self- tolerance (Fig. 1.6)41. Immune checkpoint inhibitor (CPI) antibodies liberate immune cells from the brake and reactivate anti-tumor immunity (Fig. 1.7) 42.
Fig. 1.6. The spectrum of activity of PD-1 and PD-L1 antibodies in many types of cancer defines the ‘PDLoma’ concept. PD-1: programmed cell death protein; Mel: melanoma; RCC:
renal cell carcinoma; NSCLC: non-small cell lung cancer; HNSCC: head and neck squamous cell carcinoma; NHL: non-Hodgkin lymphoma; MSI high CRC: microsatellite instability colorectal carcinoma; TNBC: triple-negative breast cancer; Mesoth: mesothelioma; HCC: hepatocellular carcinoma; oesophag: oesophageal cancer; SCLC: small cell lung cancer.
Reprinted with the permission from ref 41.
Fig. 1.7. Role of cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) (panel A) and programmed death-1 (PD-1) (panel B) in immunoregulation. Reprinted with the permission from ref 42.
1.2.1.3 Immune-stimulating anticancer small molecules
In homeostatic cell cycling, cell death often occurs as apoptosis which is considered as a tolerogenic or null event from the viewpoint of immunology 43-44. In contrast, recent reports point out that some types of anti-neoplastic agents can induce immune responses against dead-cell antigens 45-46, contributing to favorable clinical outcomes. This is called immunogenic cell death (ICD), as highlighted by secretion of calreticulin (CRT), ATP, and high-mobility group box1 (HMGB1) (Fig. 1.8)44. Mainly, these three factors efficiently activate immature dendritic cells (DCs) and facilitate the presentation of tumor-derived antigens to T cells. Two types of stresses, specifically endoplasmic reticulum stress and autophagy stress, are essential requirements for ICD induction44. Anthracyclines (such as Dox) are well known ICD-inducers. Anthracyclines increase the number of tumor infiltrating lymphocytes in some population of patients 47 and synergistically suppress tumor growth in combination with CPI antibodies 48. There are multiple clinical trials examining the combination therapies of ICD-inducers and CPIs (e.g. NCT02648477), showing the potential importance of the regimen.
Fig. 1.8. Properties of immunogenic cell death (ICD). CD91, P2X purinergic receptor 7 (P2RX7), and toll-like receptor 4 (TLR4) are receptors of CRT, ATP, and HMGB1, respectively.
Reprinted with the permission from ref 44.
1.2.2 Adverse immune responses upon cancer immunotherapy
As I explained, anti-cancer immune responses can be induced/facilitated through a variety of therapeutic approaches. However, immune responses can also be directed to normal cells and drugs, often causing severe adverse events such as unacceptable toxicity and reduction of therapeutic efficacy.
1.2.2.1 Autoimmune responses
Therapeutics capable of activating host immune system potentially cause immune related adverse events (irAEs). Generally speaking, irAEs are the consequences of the autoimmune responses against healthy tissues. In the case of CPI treatment, irAEs occur systemically through non-specific activation of immune system (Fig. 1.7) 41. Those include enterocolitis, dermatitis, hypophysitis, hepatitis, arthritis, diarrhea, rash, fatigue, nausea, pruritus etc 49-51. About 96% of melanoma patients treated with combined Nivolumab (anti-PD-1) and Ipilimumab (anti-CTLA-4) therapy experienced adverse events, and 36% of the patients discontinued the treatment due to unacceptable side effects52. Immune responses against normal cells are problematic in targeted immunotherapeutics as well, because target antigens overexpressed in cancer cells are also expressed in normal cells. Thus, innovative approaches for mitigating irAEs of cancer immunotherapeutics are strongly desired.
1.2.2.2 Production of anti-drug antibody
If therapeutic molecules are recognized as immunogenic substances by host immune system, anti- drug antibodies (ADAs) can be eventually produced. Immunogenic substances are expected to be larger than 6000 Da of molecular mass to induce crosslinking of membrane IgM on naïve B cells.
In addition, the existence of immunogenic epitopes in biopharmaceuticals leads to ADA production (Fig. 1.10, 1.11) 53-54. ADAs show negative influences on therapeutic outcomes of the drugs. For example, even fully human monoclonal antibody (mAb) against tumor necrosis factor- a (TNF-a), namely adalimumab, suffers from ADA production53, resulting in reduced serum concentration and therapeutic efficacy 55. This indicates that production of completely non- immunogenic antibodies is difficult due to the nature of antigen-recognition sites, although immunogenicity of the complementarity determining region could be partially reduced while maintaining its affinity by modification 56. Therefore, natural proteins and small molecules could be good candidates of pharmaceutics with less risks of ADA production.
Fig. 1.10. Putative immunogenic sites on anti-TNF antibody constructs. Reprinted with the permission from ref 53.
Figure 1.11. Molecular structures of biologic drugs used to treat psoriasis. Neutralizing anti- drug antibodies (ADAs) bind to murine epitopes and idiotopes located at the antigen-binding site and prevent fixation of the therapeutic agent to its target, reducing its clinical efficacy. In contrast, ADAs that bind to allotopes and human neo-antigens at the hinge of fusion proteins are usually non-neutralizing. Both neutralizing and non-neutralizing ADAs may reduce drug efficacy through the formation of immune complexes, which may lead to increased drug clearance. Reprinted with the permission from ref 54.
1.3 Overview of this thesis
Targeted drug delivery and immunotherapy are two paradigms that hold huge potential to cure patients of cancers. However, both still have some major challenges. In the case of targeted drug delivery, acquired resistances to delivery methods as well as to cargo molecules should be addressed. Non-desired immune responses against normal cells and drugs are limiting the potential of cancer immunotherapy.
In chapter 2, we sought to develop a novel protein for anti-cancer drug delivery. Collagen-binding domain (CBD) of von Willebrand factor was fused to the N-terminus of mouse serum albumin (SA) to produce CBD-SA. CBD-SA is expected to actively target leaky tumor vasculature, and passively accumulate into tumor tissues through EPR effect. Exposure of collagens in tumor vasculature is thought to be a general feature of solid tumors. Further, collagen is less likely to show structural changes by mutation because its main producer cell is fibroblast, which is reportedly have lower mutation rates than cancer cells 57-59. Thus, our hybrid approach of active and passive targeting by CBD-SA has potential to fight with acquired resistance of cancer cells to therapeutic intervention. We showed that Dox conjugated with CBD-SA (Dox-CBD-SA) enhances the therapeutic efficacy of Dox, and Dox-CBD-SA and anti-PD-1 CPI antibody synergistically eradicate a mouse model of colon carcinoma. In chapter 3, we intended to develop a new class of small molecules which enables to exploit endogenous antibodies for anti-tumor immune responses, called Fc-binding antibody-recruiting small molecules (Fc-ARMs). Fc-ARM is composed of a Fc- binding peptide and a targeting ligand, and it recruits endogenous antibodies abundant in the blood stream to induce antibody-dependent cell-mediated cytotoxicity against cancer cells. Small molecules like Fc-ARM have potential to induce effector functions of antibodies without administration of antibodies, thereby providing a new therapeutic option with less risks of irAEs.
Moreover, the Fc-ARM strategy substantially enhances the available amounts of endogenous antibodies in the blood stream, providing robust opportunities to induce antibody-mediated immune responses. This is a very important breakthrough in this field. In chapter 4, we introduced another potential application of Fc-ARM for enhancement of the selectivity of ADCC. We
selectivity of immune responses would be beneficial for reducing adverse effects of cancer immunotherapeutics.
1.4 References
1. Siegel, R. L.; Miller, K. D.; Jemal, A., Cancer statistics, 2019. CA: a cancer journal for clinicians 2019, 69 (1), 7-34.
2. Higashi, T.; Katanoda, K.; Sobue, T.; Mikami, H.; Wakao, F., Cancer statistics in Japan—
2018. 2019.
3. Verma, S.; Miles, D.; Gianni, L.; Krop, I. E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E., Trastuzumab emtansine for HER2-positive advanced breast cancer. New England Journal of Medicine 2012, 367 (19), 1783-1791.
4. Shi, J.; Kantoff, P. W.; Wooster, R.; Farokhzad, O. C., Cancer nanomedicine: progress, challenges and opportunities. Nature Reviews Cancer 2017, 17 (1), 20.
5. Hoogenboom, H. R., Selecting and screening recombinant antibody libraries. Nature biotechnology 2005, 23 (9), 1105-1116.
6. Nelson, A. L.; Dhimolea, E.; Reichert, J. M., Development trends for human monoclonal antibody therapeutics. Nature reviews drug discovery 2010, 9 (10), 767-774.
7. Frenzel, A.; Schirrmann, T.; Hust, M. In Phage display-derived human antibodies in clinical development and therapy, MAbs, Taylor & Francis: 2016; pp 1177-1194.
8. Muyldermans, S.; Baral, T.; Retamozzo, V. C.; De Baetselier, P.; De Genst, E.; Kinne, J.;
Leonhardt, H.; Magez, S.; Nguyen, V.; Revets, H.; Rothbauer, U.; Stijlemans, B.; Tillib, S.;
Wernery, U.; Wyns, L.; Hassanzadeh-Ghassabeh, G., Camelid immunoglobulins and nanobody technology. Veterinary immunology and immunopathology 2009, 128 (1-3), 178-183.
9. Oliveira, S.; Heukers, R.; Sornkom, J.; Kok, R. J.; en Henegouwen, P. M. v. B., Targeting tumors with nanobodies for cancer imaging and therapy. Journal of Controlled Release 2013, 172 (3), 607-617.
10. Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F. Y., Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications.
FEBS letters 2010, 584 (12), 2670-2680.
11. Ståhl, S.; Gräslund, T.; Karlström, A. E.; Frejd, F. Y.; Nygren, P.-Å.; Löfblom, J., Affibody molecules in biotechnological and medical applications. Trends in biotechnology 2017, 35 (8),
13. McKeague, M.; DeRosa, M. C., Challenges and opportunities for small molecule aptamer development. Journal of nucleic acids 2012, 2012.
14. Chen, K.; Liu, B.; Yu, B.; Zhong, W.; Lu, Y.; Zhang, J.; Liao, J.; Liu, J.; Pu, Y.; Qiu, L.;
Zhang, L.; Liu, H.; Tan, W., Advances in the development of aptamer drug conjugates for targeted drug delivery. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology 2017, 9 (3), e1438.
15. Lam, K. S.; Salmon, S. E.; Hersh, E. M.; Hruby, V. J.; Kazmierski, W. M.; Knapp, R. J., A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354 (6348), 82-84.
16. Elia, A. E.; Cantley, L. C.; Yaffe, M. B., Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299 (5610), 1228-1231.
17. Fujino, T.; Goto, Y.; Suga, H.; Murakami, H., Ribosomal synthesis of peptides with multiple β-amino acids. Journal of the American Chemical Society 2016, 138 (6), 1962-1969.
18. Zorzi, A.; Deyle, K.; Heinis, C., Cyclic peptide therapeutics: past, present and future.
Current opinion in chemical biology 2017, 38, 24-29.
19. Davis, B. G.; Robinson, M. A., Drug delivery systems based on sugar-macromolecule conjugates. Current Opinion in Drug Discovery and Development 2002, 5 (2), 279-288.
20. Zhang, Y.; Chan, J. W.; Moretti, A.; Uhrich, K. E., Designing polymers with sugar-based advantages for bioactive delivery applications. Journal of Controlled Release 2015, 219, 355-368.
21. LoRusso, P. M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M. X., Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2–positive cancer. Clinical Cancer Research 2011, 17 (20), 6437-6447.
22. Matsumura, Y.; Maeda, H., A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer research 1986, 46 (12 Part 1), 6387-6392.
23. Maeda, H.; Nakamura, H.; Fang, J., The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Advanced drug delivery reviews 2013, 65 (1), 71-79.
24. Koga, Y.; Manabe, S.; Aihara, Y.; Sato, R.; Tsumura, R.; Iwafuji, H.; Furuya, F.;
Antitumor effect of antitissue factor antibody‐MMAE conjugate in human pancreatic tumor xenografts. International journal of cancer 2015, 137 (6), 1457-1466.
25. Akiyama, Y.; Mori, T.; Katayama, Y.; Niidome, T., The effects of PEG grafting level and injection dose on gold nanorod biodistribution in the tumor-bearing mice. Journal of Controlled Release 2009, 139 (1), 81-84.
26. Abdalla, A. M.; Xiao, L.; Ullah, M. W.; Yu, M.; Ouyang, C.; Yang, G., Current challenges of cancer anti-angiogenic therapy and the promise of nanotherapeutics. Theranostics 2018, 8 (2), 533-548.
27. O’Brien, M. E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.;
Kieback, D.; Tomczak, P.; Ackland, S., Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX™/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Annals of oncology 2004, 15 (3), 440-449.
28. Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.;
Gallen, M.; Marsters, S.; Tsai, S. P.; Minoche, A.; Seshagiri, S.; Serrano, S.; Himmelbauer, H.;
Bellmunt, J.; Rovira, A.; Settleman, J.; Bosch, F.; Albanell, J., Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nature medicine 2012, 18 (2), 221-223.
29. Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O. C., Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Advanced drug delivery reviews 2014, 66, 2-25.
30. Danhier, F., To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? Journal of Controlled Release 2016, 244, 108-121.
31. Press release: The Nobel Prize in Physiology or Medicine 2018. The Nobel Assembly at Karolinska Institutet 2018.
32. Scott, A. M.; Wolchok, J. D.; Old, L. J., Antibody therapy of cancer. Nature reviews cancer 2012, 12 (4), 278-287.
33. Srivastava, R. M.; Lee, S. C.; Andrade Filho, P. A.; Lord, C. A.; Jie, H.-B.; Davidson, H.
34. Chen, X.; Cai, H. H., Monoclonal antibodies for Cancer therapy approved by FDA. MOJ Immunology 2016, 4 (2), 00120.
35. Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T., Induced expression of PD‐1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal 1992, 11 (11), 3887-3895.
36. Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T., Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes.
International immunology 1996, 8 (5), 765-772.
37. Vibhakar, R.; Juan, G.; Traganos, F.; Darzynkiewicz, Z.; Finger, L. R., Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Experimental cell research 1997, 232 (1), 25-28.
38. Okazaki, T.; Honjo, T., The PD-1–PD-L pathway in immunological tolerance. Trends in immunology 2006, 27 (4), 195-201.
39. Brunet, J.-F.; Denizot, F.; Luciani, M.-F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.-G.;
Golstein, P., A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328 (6127), 267-270.
40. Krummel, M. F.; Allison, J. P., CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. Journal of Experimental Medicine 1995, 182 (2), 459-465.
41. Michot, J.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.;
Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; Massard, C.; Fuerea, A.; Ribrag, V.;
Gazzah, A.; Armand, J. P.; Amellal, N.; Angevin, E.; Noel, N.; Boutros, C.; Mateus, C.; Robert, C.; Soria, J.-C.; Marabelle, A.; Lambotte, O., Immune-related adverse events with immune checkpoint blockade: a comprehensive review. European journal of cancer 2016, 54, 139-148.
42. Soularue, E.; Lepage, P.; Colombel, J. F.; Coutzac, C.; Faleck, D.; Marthey, L.; Collins, M.; Chaput, N.; Robert, C.; Carbonnel, F., Enterocolitis due to immune checkpoint inhibitors: a systematic review. Gut 2018, 67 (11), 2056-2067.
43. Green, D. R.; Ferguson, T.; Zitvogel, L.; Kroemer, G., Immunogenic and tolerogenic cell death. Nature Reviews Immunology 2009, 9 (5), 353-363.
44. Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L., Immunogenic cell death in cancer
45. Casares, N.; Pequignot, M. O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.;
Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; Coutant, F. d. r.; Métivier, D.; Pichard, E.;
Aucouturier, P.; Pierron, G. r.; Garrido, C.; Zitvogel, L.; Kroemer, G., Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. Journal of Experimental Medicine 2005, 202 (12), 1691-1701.
46. Zitvogel, L.; Kepp, O.; Kroemer, G., Immune parameters affecting the efficacy of chemotherapeutic regimens. Nature reviews Clinical oncology 2011, 8 (3), 151-160.
47. Ladoire, S.; Mignot, G.; Dabakuyo, S.; Arnould, L.; Apetoh, L.; Rébé, C.; Coudert, B.;
Martin, F.; Bizollon, M. H.; Vanoli, A.; Coutant, C.; Fumoleau, P.; Bonnetain, F.; Ghiringhelli, F., In situ immune response after neoadjuvant chemotherapy for breast cancer predicts survival. The Journal of pathology 2011, 224 (3), 389-400.
48. Rios-Doria, J.; Durham, N.; Wetzel, L.; Rothstein, R.; Chesebrough, J.; Holoweckyj, N.;
Zhao, W.; Leow, C. C.; Hollingsworth, R., Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia 2015, 17 (8), 661-670.
49. Maker, A. V.; Phan, G. Q.; Attia, P.; Yang, J. C.; Sherry, R. M.; Topalian, S. L.; Kammula, U. S.; Royal, R. E.; Haworth, L. R.; Levy, C.; Kleiner, D.; Mavroukakis, S. A.; Yellin, M.;
Rosenberg, S. A., Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte–associated antigen 4 blockade and interleukin 2: a phase I/II study. Annals of surgical oncology 2005, 12 (12), 1005-1016.
50. Phan, G. Q.; Yang, J. C.; Sherry, R. M.; Hwu, P.; Topalian, S. L.; Schwartzentruber, D. J.;
Restifo, N. P.; Haworth, L. R.; Seipp, C. A.; Freezer, L. J.; Morton, K. E.; Mavroukakis, S. A.;
Duray, P. H.; Steinberg, S. M.; Allison, J. P.; Davis, T. A.; Rosenberg, S. A., Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proceedings of the National Academy of Sciences 2003, 100 (14), 8372-8377.
51. Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F. L.; Carbonnel, F.;
Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; Lanoy, E.; Texier, M.; Libenciuc, C.;
Eggermont, A. M. M.; Soria, J.-C.; Mateus, C.; Caroline, R., Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nature reviews Clinical oncology 2016, 13 (8),
52. Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J. J.; Cowey, C. L.; Lao, C. D.;
Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; Ferrucci, P. F.; Hill, A.; Wagstaff, J.;
Carlino, M. S.; Haanen, J. B.; Maio, M.; Marquez-Rodas, I.; McArthur, G. A.; Ascierto, P. A.;
Long, G. V.; Callahan, M. K.; Postow, M. A.; Grossmann, K.; Sznol, M.; Dreno, B.; Bastholt, L.;
Yang, A.; Rollin, L. M.; Horak, C.; Hodi, F. S.; Wolchok, J. D., Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New England journal of medicine 2015, 373 (1), 23-34.
53. Bendtzen, K., Immunogenicity of anti-TNF-α biotherapies: II. Clinical relevance of methods used for anti-drug antibody detection. Frontiers in immunology 2015, 6, 109.
54. Jullien, D.; Prinz, J. C.; Nestle, F. O., Immunogenicity of biotherapy used in psoriasis: the science behind the scenes. Journal of Investigative Dermatology 2015, 135 (1), 31-38.
55. Bartelds, G. M.; Krieckaert, C. L.; Nurmohamed, M. T.; van Schouwenburg, P. A.; Lems, W. F.; Twisk, J. W.; Dijkmans, B. A.; Aarden, L.; Wolbink, G. J., Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. Jama 2011, 305 (14), 1460-1468.
56. Harding, F. A.; Stickler, M. M.; Razo, J.; DuBridge, R. In The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions, MAbs, Taylor & Francis: 2010; pp 256-265.
57. Walter, K.; Omura, N.; Hong, S.-M.; Griffith, M.; Goggins, M., Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer biology & therapy 2008, 7 (6), 882-888.
58. Qiu, W.; Hu, M.; Sridhar, A.; Opeskin, K.; Fox, S.; Shipitsin, M.; Trivett, M.; Thompson, E. R.; Ramakrishna, M.; Gorringe, K. L.; Polyak, K.; Haviv, I.; Campbell, I. G., No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nature genetics 2008, 40 (5), 650-655.
59. Hosein, A. N.; Wu, M.; Arcand, S. L.; Lavallée, S.; Hébert, J.; Tonin, P. N.; Basik, M., Breast carcinoma–associated fibroblasts rarely contain p53 mutations or chromosomal aberrations.
Cancer research 2010, 70 (14), 5770-5777.
Chapter 2; Collagen-binding serum albumin as a drug conjugate carrier for treating solid tumors
2.1 Introduction
In this chapter, we sought to enhance the utility of serum albumin (SA) as a drug carrier protein for cancer therapy through fusion of collagen binding domain (CBD) to SA. Firstly, I introduced some researches aiming for extracellular matrix targeted drug delivery as a potential solution of resistance to targeted drug delivery due to genetic instability and heterogeneity of cancer cells in solid tumors. Next, main contents of our project were explained.
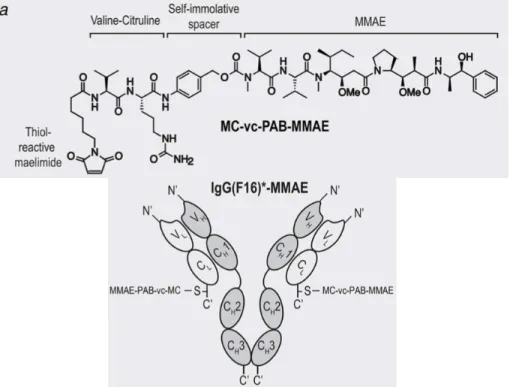
Extracellular matrix (ECM) is a non-cellular constituent of tissue which is essential for structural support for cells as well as regulation of physiological signaling of cells. ECM exists ubiquitously in our tissues, but only exposed to the blood stream in fenestrated blood vessels or abnormally permeable tumor blood vessels. Although a degree of fenestration and a density of ECM in each tumor may be different, exposed ECM would be one of the general features of tumor microenvironment. In addition, it can be expected that ECM targeting is less likely to induce down- regulation of binding due to mutation because the strategy does not target genetically instable tumor cells. Indeed, it is experimentally suggested that cancer-associated fibroblasts (main producers of ECM) are genetically stable1-4. Thus, ECM could be a promising group of candidate molecules for active targeting to fight with cancer heterogeneity and drug resistance. Main components of ECM are collagen, elastin, laminin, fibrin, etc5. Previously, antibodies against fibronectin6, type IV collagen7, fibrin8, and tenascin-C (Fig. 2.1)9-10 were developed as drug conjugate carriers to deliver imaging agents or anti-cancer therapeutics. As suggested by Matsumura et al in 2011, targeted delivery of anti-cancer agents to ECM can turn cancer-stroma into a reservoir, from where drugs are released and diffuse throughout the tumor tissue in sustained manner (Fig. 2.2)7. This concept is in contrast to conventional antibody-drug conjugates (ADCs) which aim to induce cellular uptake of the conjugates upon binding to target proteins, and
Fig. 2.1. Chemical structure of the MC-vc-PAB-MMAE drug and schematic representation of IgG(F16)-MMAE.
MC; maleimidocaprioyl, vc; valine-citruline, PAB; para-aminobenzylcarbonyl, MMAE; mono- methyl auristatin E, F16; a clone specific to a splice isoform of tenascin-C. Modified and reprinted with the permission from ref10.
Serum albumin (SA) is the most abundant protein in the blood stream14. A variety of compounds including small molecules, peptides, and cytokines have been fused to, conjugated to, or formulated with SA for improved drug delivery to disease lesions. SA-binding fatty acid–modified insulin analog15, SA-fused interferon-a (IFN-a)16, and SA nanoparticle–formulated paclitaxel (Abraxane) have been developed17. In these cases, the exceptionally long plasma half-life and/or hydrophilicity of SA improves pharmacokinetics, safety, and efficacy of the drugs14. Moreover, SA can target tumors through the pathological permeability of the tumor vasculature (passive targeting)18, which is an advantage of SA-based drug delivery methods for cancer therapy.
On the basis of the rationale that combination of passive and active targeting is beneficial in tumor drug delivery19, molecular engineering approaches aiming for further improvement of SA-based drugs have been explored, such as conjugation of the targeting ligands cyclic arginylglycylaspartic acid peptide20 or mannose-6-phosphate to SA21. However, superior antitumor efficacy of modified SA as a drug carrier has not been shown compared to drugs associated with unmodified SA.
Recently, we have reported the targeted delivery of checkpoint inhibitor (CPI) antibodies and the cytokine interleukin-2 (IL-2) using a collagen-binding domain (CBD) of the A3 domain of von Willebrand factor (VWF) (Fig. 2.3)22. The A3 domain of VWF is the strongest binder to collagen type I and type III among reported nonbacterial origin proteins/peptides23. Collagens are not accessible in most tissues due to the low permeability of the vascular structure, yet are abnormally exposed to the bloodstream in the tumor microenvironment due to the enhanced permeability of the tumor vasculature19. Thus, collagens would be useful targets for active drug delivery to cancer.
In addition, collagens are overexpressed in multiple types of cancers24-25. CBD-CPI and CBD–
interleukin-2 (IL-2) showed significantly stronger antitumor effects compared with their unmodified forms in multiple murine cancer models22. In addition, treatment-related adverse events of CPI and IL-2 were greatly suppressed by CBD modification, which shows the usefulness of the CBD-based tumor-targeting strategy22. Here, we hypothesized that the CBD would be compatible with SA-based drug delivery methods, because both CBD and SA target abnormally permeable tumor vasculature, and CBD fusion adds active targeting capability to SA.
Fig. 2.3. CBD-conjugated CPI antibody and CBD-fused IL-2.
(A) Schematic of conjugation of a CBD, the recombinant von Willebrand factor A3 domain, to CPI antibody resulting in affinity for collagen. CBD–IL-2 was recombinantly expressed, with the CBD on the N terminus of IL-2, using a (GGGS)2 linker. (B) KD values of CBD–aPD-L1 and unmodified aPD-L1, aCTLA4, and IL-2 against type I and III collagen, recombinant mouse (rm) CTLA4, rmPD-L1, and/or rmIL-2Ra were measured by enzyme-linked immunosorbent assay (ELISA). N.D., not determined because of low signals. (C) 5 × 105 B16F10 cells were inoculated on day 0. CBD- or unmodified aCTLA4 and aPD-L1 (100 µg each/injection) were injected i.v.
on day 4 and 7. Histologic liver and lung sections on day 8. Black arrows indicate the sites of leukocyte infiltration. Scale bar = 200 µm. (D, E) 5 × 105 MMTV-PyMT cells were inoculated on the right mammary fat pad on day 0. CBD-aCTLA4 + CBD–aPD-L1 (CBD-CPI), aCTLA4 + aPD-L1 (CPI), or PBS was administered on day 7. Graphs depict (D) tumor volume until the first mouse died and (E) survival rate. Reprinted with the permission from ref 22.
Doxorubicin (Dox) is a small-molecule anticancer drug that is approved for treating a variety of cancers by the U.S. Food and Drug Administration. Dox internalizes within cells through transmembrane diffusion and inhibit DNA functions, leading to death of proliferating cells.
Although Dox administration extends survival of some populations of patients, antitumor efficacy is not notable partially due to acquired drug resistance. The poor therapeutic index of Dox also limits its therapeutic use. Considerable toxicity of Dox has been reported, including bone marrow suppression, excessive inflammation, and cardiotoxicity26-27. To improve efficacy, Dox is often used in combination with other chemotherapies. Recently, Dox has been reported to facilitate immune cell infiltration into tumors by inducing immunogenic cell death (ICD)28, showing the possibility of synergizing in combination treatment with immunotherapeutics such as CPI29. Other approaches to improving efficacy and maximum tolerated dose of Dox are liposomal formulation (Doxil)30 and use of a maleimide derivative of Dox with a pH-responsive cleavable linker (aldoxorubicin), which was developed to achieve in situ conjugation with cysteine-34 (in the human sequence) of endogenous SA in patients31. Aldoxorubicin shows extended blood half-life and accumulation within tumors through passive tumor targeting. The low pH within the tumor tissue (reportedly pH 6.5) allows Dox release from the conjugate with SA. Aldoxorubicin showed improved maximum tolerated dose and efficacy in mouse cancer models31-32 and in a clinical trial33 compared with unmodified Dox.
Here, we designed recombinant mouse SA (CBD-SA) in which the N terminus is fused with the C terminus of the VWF A3 domain, and aldoxorubicin was conjugated to CBD-SA via a pH- dependent cleavable hydrazone linkage34 before injection (namely, Dox-CBD-SA). We evaluated engineered CBD-SA as a tumor-targeted drug carrier protein, leading to improved antitumor efficacy and reduced toxicity by efficient Dox delivery to the tumor vasculature.
2.2 Results
2.2.1 CBD-SA binds to collagen and can be conjugated with Dox
We synthesized Dox-CBD-SA conjugates to target the tumor microenvironment (Fig. 2.4 and 2.5A). We first tested the binding abilities of CBD-SA to recombinant collagen protein in vitro.
SA was expressed recombinantly with the CBD on the N terminus of mouse SA using a (GGGS)2
linker (table S2.1). The molecular mass of CBD-SA was analyzed by matrix-assisted laser desorption/ ionization–time-of-flight mass spectrometry (MALDI-TOF MS) (fig. S2.1). We observed strong binding affinities [nanomolar range dissociation constant (Kd) values] of CBD- SA to collagen type I and type III (Fig. 2.5B and fig. S2.2). For Dox conjugation, we first converted the primary amines of the lysine residues of CBD-SA to thiols using 2-iminothiolane (also known as Traut’s reagent). Then, CBD-SA was conjugated with aldoxorubicin. Unmodified SA was also conjugated with aldoxorubicin in the same way (Dox-SA). SDS–polyacrylamide gel electrophoresis (PAGE) under non-reducing conditions revealed that Dox-SA and Dox-CBD-SA are monomeric after purification (fig. S2.3). The hydrodynamic radius of CBD-SA was also measured before and after Dox conjugation (fig. S2.4). The results also showed that CBD-SA exists in a monomeric form, and Dox conjugation did not alter this character even after a lyophilization/reconstitution cycle. Approximately three Dox molecules were modified per SA molecule and per CBD-SA molecule (Fig. 2.5C). Importantly, our conjugation method would not deteriorate the binding ability of CBD-SA to collagens because no cysteine or lysine residues are located at the binding interface between the VWF A3 domain and human collagen III (Protein Data Bank: 4DMU; fig. S2.5)35. The interface is also far from the C-terminal fusion site of the CBD to the SA domain.
Fig. 2.4. Schematic of CBD-SA mediated drug delivery to tumor.
Fig. 2.5. (A) Synthesis scheme of Dox-CBD-SA. (B) Affinities [dissociation constant (Kd) values
H N
NH2+ Cl- S O
OH NH2
OH O OH
O N
O
OH O
HO NH
N O
O O
CBD-SA
H N
NH2+ Cl- SH
CBD-SA
NH2
CBD-SA
S NH2+ Cl- Aldoxorubicin
Kd(nM) Collagen I Collagen III
CBD-SA 1.8 40.6
SA N.D. N.D.
Dox : protein ratio
Dox-SA 3.1 ± 0.1
Dox-CBD-SA 3.4 ± 0.1
C B A
Dox conjugation ratio per protein is shown. Values were calculated on the basis of the results of bicinchoninic acid assay protein quantification assay (proteins) and absorbance at 495 nm (Dox) (mean ± SD of three experimental replicates).
2.2.2 Dox release under acidic pH conditions
We tested the release profiles of Dox from conjugates under different pH conditions (Fig. 2.6).
After 48 hours of incubation, Dox was completely released from Dox-CBD-SA at pH 5.0 and 6.5 (reported tumor microenvironment condition). In contrast, only about 20% of Dox was released at pH 7.4 after 48 hours. Dox-SA showed similar release kinetics of Dox (fig. S2.6). These data demonstrate the pH-dependent release of Dox from the conjugates, consistent with previously reported release kinetics of small chemicals linked via a hydrazone bond34.
Fig. 2.6. Dox release kinetics from Dox-CBD-SA under three different pH conditions.
The amount of Dox was quantified by measuring fluorescence (excitation at 495 nm, emission at 590 nm) (n = 3, mean ± SD; two experimental replicates).
0 20 40 60
0 50 100 150
Time (h)
% Drug release
Dox-CBD-SA pH 5.0
pH 6.5 pH 7.4
2.2.3 Dox conjugates are taken up by cancer cells and retain cytotoxicity
We observed the intracellular localization of Dox conjugates and free Dox using confocal laser scanning microscopy by detecting the fluorescence of Dox. Because Dox is a major drug for treating breast cancer36, here we chose mouse mammary tumor virus-polyomavirus middle T antigen (MMTV-PyMT) murine breast cancer as an experimental model. The MMTV-PyMT cells were seeded and incubated overnight, then cultured in the presence of Dox or Dox conjugates, and their intracellular localization was evaluated (Fig. 2.7A). After 1 hour of incubation, free Dox was detected in the cytoplasm, intracellular acidic organelles (stained by Lysotracker), and preferentially in the nucleus, indicating that its delivery is mediated by passive transmembrane diffusion. In contrast, 1 hour after addition of either Dox-SA or Dox-CBD-SA, the cytoplasm did not show strong fluorescence compared with the unconjugated Dox. Rather, punctate fluorescence was observed, with some puncta colocalized with lysosomes, indicating that Dox-SA and Dox- CBD-SA were both internalized through endocytosis pathway. 24 hours after the addition of Dox conjugates, Dox-derived fluorescence was observed in the nucleus as well, suggesting that the acidic pH in intracellular organelles induced drug liberation from the conjugates. We next tested the cytotoxicity of Dox and the conjugates in vitro. MMTV-PyMT cells or MC38 colon carcinoma cells were seeded and incubated in the presence of the Dox forms for 3 days. Cell viability tests showed that free Dox and the conjugates have comparable cytotoxicity in vitro (Fig. 2.7B and C).
Fig.2.7. Cellular uptake and cytotoxicity of the Dox conjugates.
(A) MMTV-PyMT cells were seeded and cultured overnight. Dox, Dox-SA, or Dox-CBD-SA was added (red). Before observation by confocal laser scanning microscopy, cells were also stained with LysoTracker (green). Scale bars, 20 µm. Representative pictures are presented. Two experimental replicates. (B and C) Cytotoxicity of Dox and the conjugates against MMTV-PyMT cells or MC38 cells in vitro (n = 6, mean ± SEM). Two experimental replicates. IC50, half maximal inhibitory concentration.
B C
10-8 10-7 10-6 10-5 0
50 100
Doxorubicin conc. (M-1)
% Viability
MMTV-PyMT breast cancer
Aldoxorubicin Dox-SA Dox-CBD-SA
0.29 0.19 0.15 IC50 (µM)
10-8 10-7 10-6 10-5 0
50 100
Doxorubicin conc. (M-1)
% Viability
MC38 colon carcinoma
Dox-CBD-SA Aldoxorubicin 0.70
1.80 IC50 (µM)
A
Doxorubicin Dox-SA Dox-CBD-SA1 h
24 h
Dox Lysotracker
2.2.4 Dox-CBD-SA shows similar blood plasma pharmacokinetics as aldoxorubicin and efficiently accumulates in tumors
Aldoxorubicin reacts with endogenous SA rapidly after systemic administration; therefore it possesses substantially long blood circulation time as a small molecule14, 31-32. We tested the plasma pharmacokinetics of aldoxorubicin with or without prior conjugation of SA and CBD-SA using tumor-free FVB mice. Blood plasma half-lives of aldoxorubicin, Dox-SA, and Dox-CBD- SA were found to be similar (Fig. 2.8A and B). We also examined the plasma pharmacokinetics of SA and CBD-SA labeled with fluorescent probes with a pH-insensitive linker (fig. S2.7). The result showed that the half-lives of each protein conjugated with either Dox or dye were similar, suggesting that liberation rates of Dox from the conjugates are very slow in the blood stream. We next hypothesized that CBD fusion to SA would enhance the amount of Dox within the tumor via active targeting against collagens within the tumor vasculature. To test this hypothesis, we measured the amounts of Dox within MMTV-PyMT tumor after a single intravenous administration. Dox-CBD-SA showed significantly higher tumor accumulation of Dox compared with aldoxorubicin and Dox-SA at 2 hours after injection (Fig. 2.8C). Conjugation with CBD-SA achieved the highest tumor accumulation of Dox after 24 hours of injection as well, showing a significant increase compared with aldoxorubicin. Histological analysis after intravenous injection of fluorescently labeled CBD-SA or SA revealed that CBD-SA colocalized with CD31 staining within tumor tissue, showing that CBD-SA targets the leaky tumor vasculature (Fig. 2.8D). These data demonstrate that CBD fusion to SA to which Dox is conjugated enables Dox to target tumors, resulting in enhanced tumor accumulation of Dox.
Fig. 2.8. Dox-CBD-SA shows comparable plasma pharmacokinetics with Dox-SA and higher tumor accumulation than aldoxorubicin and Dox-SA.
(A) Aldoxorubicin, Dox-SA, or Dox-CBD-SA (5 mg/kg on a Dox basis) was injected to tumor- free FVB mice via tail vein administration. Blood plasma was collected at the indicated time points.
Dox concentration in plasma was measured by fluorescence (mean ± SEM; n = 4 for aldoxorubicin, n = 5 for Dox-SA and Dox-CBD-SA). (B) Plasma half-lives of Dox were calculated using two- phase exponential decay: MFI (t) = Ae−at + Be−bt. t1⁄2, a, fast clearance half-life; t1⁄2, b, slow clearance half-life (mean ± SEM; n = 4 for aldoxorubicin, n = 5 for Dox-SA and Dox-CBD-SA).
(C) MMTV-PyMT tumor-bearing mice were treated with aldoxorubicin, Dox-SA, or Dox-CBD- SA (4.16 mg/kg on a Dox basis). Tumors were harvested 2 hours or 24 hours after injection, and the amount of Dox within the tumors was quantified (mean ± SEM; n = 5 for 2 hours, n = 7 for 24 hours per group). (D) DyLight 488–labeled SA (100 µg) or equimolar amounts of DyLight 488–
labeled CBD-SA were injected to MMTV-PyMT tumor-bearing mice via tail vein administration.
A B
t½, α(h) t½, β(h) Aldoxorubicin 4.0×10-2 ± 2.8×10-3 22.6 ± 3.3
Dox-SA 5.0×10-2± 7.8×10-3 8.4 ± 1.7 Dox-CBD-SA 9.0×10-2± 2.4×10-2 6.4 ± 1.3
C D
2h 24h
0 2 4 6
%ID/g
Tumor
Aldoxorubicin Dox-SA Dox-CBD-SA
***
*
N.S. N.S.
0 20 40 60 80
0.1 1 10 100
Time after injection (h)
Injected dose (%)
Blood plasma Aldoxorubicin Dox-SA Dox-CBD-SA
CD31 SA DAPI
CD31 CBD-SA DAPI
antibody. Scale bars, 100 µm. Representative images of three tumors were shown in each condition.
Two experimental replicates. Statistical analyses were done using analysis of variance (ANOVA) with Tukey’s test. *P < 0.05; **P < 0.01; N.S., not significant.
2.2.5 Dox-CBD-SA demonstrates enhanced antitumor efficacy in the MMTV-PyMT murine breast cancer model
Motivated by the plasma half-life and tumor accumulation studies, we evaluated the antitumor effects of Dox-CBD-SA in vivo. MMTV-PyMT orthotopic tumor-bearing mice received a single administration of Dox or the Dox conjugates (5 mg/kg on a Dox basis) via the tail vein. Dox-SA and Dox-CBD-SA significantly suppressed tumor growth compared with untreated group, whereas aldoxorubicin did not (Fig. 2.9A and 2.9C to F). This result suggests that conjugation of Dox with SA prior to injection would provide a higher therapeutic effect than in situ conjugation of aldoxorubicin with endogenous SA. Notably, Dox-CBD-SA showed a greater therapeutic effect compared with Dox-SA. Dox-CBD-SA therapy significantly extended the survival of mice compared with all the other groups (Fig. 2.9B) and induced complete tumor remission in 2 of 12 mice. These data demonstrate that CBD-fused SA functions as a superior Dox carrier compared to unmodified SA in terms of antitumor efficacy in MMTV-PyMT breast cancer model.