Control of Stereospecificity in the Radical Polymerization of
N-Methyl-N-(2-pyridyl)acrylamide by Conformational Switching of the Monomer
with Protonation
Tomohiro Hirano, Akihiro Morikami, Koichi Ute
Department of Chemical Science and Technology, Institute of Technology and Science, The University of Tokushima, 2-1 Minamijosanjima, Tokushima 770-8506, Japan
Correspondence to : T. Hirano (E-mail: hirano@chem.tokushima-u.ac.jp)
ABSTRACT: Radical polymerization of N-methyl-N-(2-pyridyl)acrylamide (MPyAAm) was carried out in dichloromethane at low temperatures in the presence of trifluoroacetic acid (TFA). The m dyad contents of the polymers obtained at 0°C increased linearly from 37% to 60% with increase in the [TFA]0/[MPyAAm]0 ratio from
unity to 5. NMR analysis of MPyAAm-TFA mixtures in dichloromethane-d2 revealed
that the favorable conformation in terms of the pyridyl group to the carbonyl group in MPyAAm switched from s-trans to s-cis by protonation. The results suggest that controlling the conformation of MPyAAm resulted in control of the stereospecificity in radical polymerization of the monomer.
Keywords: radical polymerization; stereospecific polymerization; isotactic;
This is the peer reviewed version of the following article: Hirano, T., Morikami, A. and Ute, K. (2011), Control of stereospecificity in the radical polymerization of N‐ methyl‐N‐(2‐pyridyl)acrylamide by conformational switching of the monomer with protonation. J. Polym. Sci. A Polym. Chem., 49: 4164-4171., which has been published in final form at https://doi.org/10.1002/pola.24858. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Use of Self-Archived Versions.
syndiotactic; N-methyl-N-(2-pyridyl)acrylamide; monomer conformation;
Running Head: Stereospecific radical polymerization
INTRODUCTION
Radical polymerization of N-monosubstituted acrylamide generally gives atactic polymer. To prepare stereoregular polymers of N-monosubstituted acrylamide, it is necessary to use Lewis acids,1,2 Lewis bases,3,4 alkyl alcohols5,6 or fluorinated
alcohols7,8 as an additive to the polymerization mixture. On the other hand, radical
polymerization of N,N-disubstituted acrylamide affords stereoregular polymer without an additive. The stereospecificity in the polymerization depends on the structure of the N-substituent in the monomer; radical polymerization of N,N-dimethylacrylamide (DMAAm) provides isotactic polymer, whereas polymerization of N,N-diphenylacrylamide (DPhAAm) gives syndiotactic polymer.9
Recently we found that radical polymerization of N-methyl-N-phenylacrylamide (MPhAAm) gave syndiotactic polymer.10 According to
the conformational analysis of N-aryl-N-methylamide,11-15 an N-aryl substituent favors
the form s-trans to the carbonyl group and perpendicular to the planar amide group. This suggests that the N-substituent s-trans to the carbonyl group of N,N-disubstituted acrylamide should play a determining role in the stereospecificity of the radical polymerization.
Monomer conformation can be one of important factors in the stereospecificity of radical polymerization. In fact, it is known that monomer conformation affects the stereochemistry in the propagating reaction in the radical polymerization of 1,1-disubstituted vinyl monomers with an optically active ester group, near the ceiling temperature.16 Thus, it is assumed that stereospecificity of radical
polymerization of N-aryl-N-methylacrylamides can be controlled by strategically controlling the monomer conformation, even when achiral monomers are used.
Recently, it was reported that N-methyl-N-(2-pyridyl)amides switched their conformation from s-trans to s-cis O=C-N-Py with protonation.17,18 If
N-methyl-N-(2-pyridyl)acrylamide (MPyAAm) undergoes similar conformational switching, addition of acid to the radical polymerization of MPyAAm is expected to change the stereospecificity from syndiotactic to isotactic. In this paper, we report the effect of trifluoroacetic acid (TFA) and methanesulfonic acid (MSA) on the stereospecificity of radical polymerization of MPyAAm. Conformational switching of MPyAAm with protonation is also discussed on the basis of NMR analysis of mixtures
of MPyAAm and TFA.
EXPERIMENTAL Materials
MPhAAm was prepared according to literature methods.10 Dimethyl
2,2’-azobisisobutyrate (MAIB) (supplied by Otsuka Chemical Co., Ltd, Japan) was recrystallized from methanol. Acryloyl chloride, triethylamine, anhydrous dichloromethane, anhydrous tetrahydrofuran (THF), anhydrous acetonitrile, HCl aqueous solution (12 N) (Kanto Chemical Co., Inc., Japan), trifluoroacetic acid (TFA), methanesulfonic acid (MSA), 2-(methylamino)pyridine (Tokyo Chemical Industry Co., Ltd, Japan), diethyl ether, methanol, and ethyl acetate (Kishida Chemical Co., Ltd, Japan) were used without further purification.
Synthesis of MPyAAm
To a stirred solution of acryloyl chloride (36.2 g, 0.40 mol) in THF (300 mL) were added 2-(methylamino)pyridine (21.6 g, 0.20 mol) and triethylamine (40.5 g, 0.40 mol) in THF (150 ml) dropwise at 3-5°C. After stirring the mixture for 24 h at room temperature, the solid precipitate that had formed was removed by filtration, and the solvent was evaporated. The residue was chromatographed (silica gel, ethyl acetate) to give 14.9 g of MPyAAm (46 %): red orange oil; 1H NMR (400 MHz, CDCl3 at 35°C), δ
8.51 (m, 1H), 7.74 (m, 1H), 7.21 (m, 1H), 7.19 (m, 1H), 6.41 (dd, 1H, 2J = 2.0 Hz, 3J =
Hz), 3.46 (s, 3H); 13C NMR (100 MHz, CDCl3 at 35°C), δ 166.22, 155.90, 149.23,
138.16, 129.27, 127.89, 121.76, 120.92, 35.36. Anal. Calc. for C9H10NO: C, 66.64; H,
6.22; N, 17.28. Found: C, 66.38; H, 6.13; N, 17.22.
Polymerization
A typical polymerization procedure was as follows. MPyAAm (1.0948 g, 6.75 mmol) was dissolved in CH2Cl2 to prepare 3 mL of solution, and TFA (0.7697 g, 6.75 mmol)
was dissolved in CH2Cl2 to prepare 3 mL of solution. Two milliliters of each solution
were transferred to a glass ampoule cooled to 0°C. MAIB (0.0104 g, 4.52×10–2 mmol)
was dissolved in CH2Cl2 to prepare 1 mL of solution and 0.5 mL of this solution was
added to the mixture of MPyAAm and TFA in CH2Cl2 at 0°C, giving final
concentrations: [MPyAAm]0=1.0 mol L–1, [TFA]0=1.0 mol L–1, [MAIB]0=5.0×10–3 mol
L–1. The glass ampoule was degassed and filled with nitrogen three times. The mixture
was irradiated at a distance of ~5cm from an UV-LED lamp (375nm, Optocode Co., Japan) to initiate polymerization. After 16 h, the polymerization mixture was poured into diethyl ether (500 mL). The precipitated polymer was collected by centrifugation, and dried in vacuo. The polymer yield was determined gravimetrically.
Measurements
1H NMR spectra of the polymers were obtained using an EX-400 spectrometer (JEOL
Ltd.). The tacticity of the polymers was determined from the 1H NMR signals of the
(DMSO-d6) at 150°C. 1H and 13C NMR spectra of MPyAAm monomer in the presence
or absence of TFA were measured in CD2Cl2 at 0°C with an ECX-400 spectrometer
(JEOL Ltd.) operated at 400 MHz for 1H and 100 MHz for 13C.
The molecular weights and molecular weight distributions of the polymers were determined by size exclusion chromatography (SEC), using polystyrene samples as molecular weight standards. SEC was performed with an HLC 8220 chromatograph (Tosoh Co.) equipped with TSK gel columns (SuperHM-M (6.5 mm ID×150 mm) and SuperHM-H (6.5 mm ID×150 mm), Tosoh Co.) and UV detector (UV-8220, Tosoh Co.). Dimethylformamide containing LiBr (10 mmol L-1) was used as eluent at 40°C with
flow rate 0.35 mL min-1. The initial polymer concentration was 1.0 mg mL-1.
No peaks were observed in the chromatograms of poly(MPyAAm)s, probably because polymers containing pyridyl groups were absorbed by the stationary phase. Thus, the molecular weights of poly(MPyAAm)s were measured after poly(MPyAAm)s were converted into hydrochloride salts.
Preparation of poly(MPyAAm) hydrochloride
Poly(MPyAAm) (ca. 30 mg, 0.19 mmol of monomeric unit) was dissolved in 2 mL of HCl methanol solution (2.0 N, 4.0 mmol of HCl), and 4 mL of diethyl ether was added dropwise to the solution. After removal of the supernatant solution by decantation, the precipitated polymer was dried in vacuo. Signals of the pyridinium protons were not observed in the 1H NMR spectrum of the precipitated polymer
signals of the pyridyl group exhibited a slight down-field shift after treatment with HCl, suggesting that the pyridyl groups in poly(MPyAAm)s were converted into HCl salts.
RESULTS AND DISCUSSION
Radical Polymerization of MPyAAm in CH3CN at 0 or –40°C in the Presence of
TFA or MSA
Radical polymerization of MPyAAm was carried out in CH3CN at 0°C in the
absence of acids; however, no polymer was obtained. The polymerization was then carried out in CH3CN in the presence of TFA (Table 1, runs 1-3), and polymers were
obtained in moderate yields. The fact that hydrogen bonding formation between C=O and –OH significantly enhances kp in radical polymerization of α,β-unsaturated ester
monomers,19-22 suggested that at least the carbonyl group in MPyAAm was protonated,
resulting in formation of polymers.
Polymer with m dyad content 38% was obtained in the presence of an equimolar amount of TFA. This value (38%) is comparable to the m dyad content (34%) of poly(MPhAAm) prepared in CH2Cl2 at 0°C (cf. Table 3), suggesting that MPyAAm
essentially favored the s-trans O=C-N-Py conformation and gave syndiotactic-rich polymers by a mechanism similar to that proposed for the MPhAAm polymerization10.
The m dyad content increased to 44% with increase in the amount of added TFA. However, isotactic specificity was not induced even in the presence of a 5-fold amount of TFA, suggesting that only partial conformational switching of MPyAAm occurred in
the presence of excess amounts of TFA at 0°C. Thus, the polymerization temperature was reduced to –40°C, in anticipation of an enhanced effect of TFA (Table 1, runs 4-6). A slight increase in the m dyad content of poly(MPyAAm) was observed regardless of the amount of added TFA, though the m dyad content did not exceed 50%.
<Table 1>
The effect of MSA (pKa: 1.6 in DMSO23), which is a stronger acid than TFA
(pKa: 3.45 in DMSO23), was then examined (Table 1, runs 7-12). The m dyad content
reached 49% in the presence of excess amounts of MSA at 0°C. By lowering the temperature to –40°C, however, the m dyad content decreased in the presence of excess MSA.
Radical Polymerization of MPyAAm in CH2Cl2 at Low Temperatures in the
Presence of TFA
The radical polymerization of MPyAAm was carried out in CH2Cl2, a less
polar solvent than CH3CN, at 0°C in the presence or absence of TFA. As for
polymerization in CH3CN, no polymer was obtained in the absence of TFA and
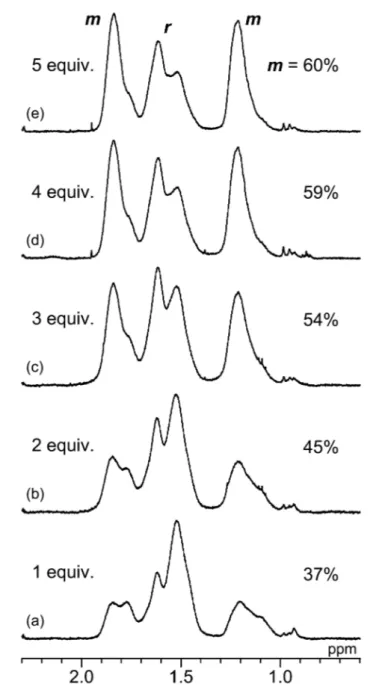
polymers were obtained in moderate yields in the presence of TFA (Table 2, runs 1-5). The addition of TFA significantly affected the stereoregularity of the poly(MPyAAm) obtained. The m dyad content of the polymer increased linearly with the amount of added TFA and reached 60%, although 4-fold and 5-fold amounts of TFA showed
similar effects (Figure 1). The value (m=60 %) is comparable with the m dyad content (62%) of poly(DMAAm) prepared in toluene at 0°C.24 This result suggested that
MPyAAm favored the s-cis O=C-N-Py conformation in CH2Cl2 in the presence of
excess amounts of TFA, and gave isotactic-rich polymers by a mechanism similar to that proposed for DMAAm polymerization24.
<Table 2> <Figure 1>
To confirm the involvement of the pyridyl group in the induction of isotactic specificity by TFA, radical polymerization of MPhAAm was carried out under the same conditions (Table 3). The addition of TFA scarcely influenced the stereoregularities of the polymers obtained, although slight decreases in the m dyad contents of the polymers obtained in the presence of equimolar and 2-fold amounts of TFA were observed. Thus, it is further suggested that the conformational switching of MPyAAm by protonation was responsible for the acid-induced isotactic specific polymerization, as expected.
<Table 3>
To examine the effect of the polymerization temperature on isotactic specificity, MPyAAm polymerizations in CH2Cl2 were carried out at –60°C to –20°C
but excess amounts of TFA were required to significantly induce isotactic specificity even at –60°C (Figure 2). The conformational switching of MPyAAm by protonation is assumed to proceed via multi-step and not single-step processes, as discussed later.
<Figure 2>
The tendency of increase in number-averaged molecular weight (Mn) of
poly(MPyAAm) with increase in the amount of the added TFA was observed, although the addition of 5-fold amount of TFA decreased Mn compared with 4-fold amount of
TFA. This tendency implies that polymerization was accelerated by protonation of the carbonyl group in MPyAAm as mentioned above and/or the rate of syndiotactic propagation differs from the rate of isotactic propagation, although the Mns determined
by SEC of poly(MPyAAm) hydrochlorides are not accurate enough to be discussed quantitatively.
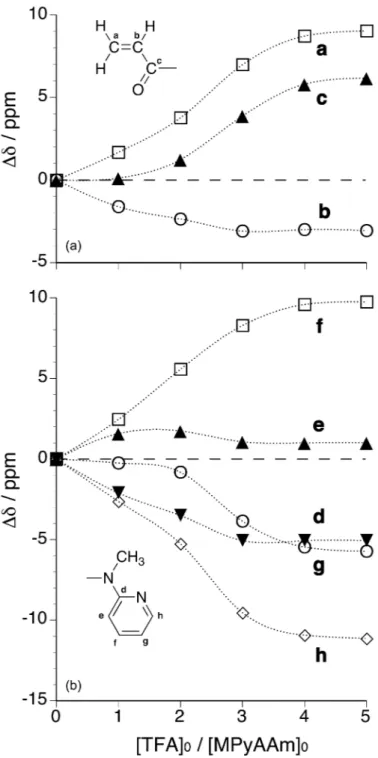
NMR Analysis of Conformational Switching of MPyAAm
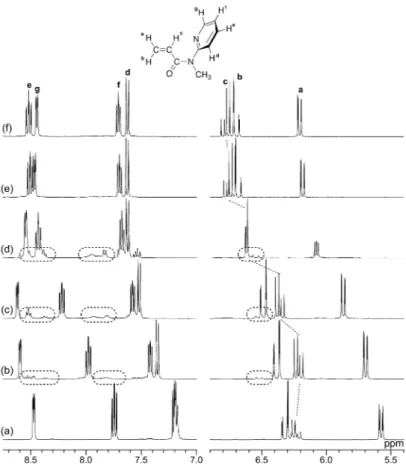
To further investigate the conformational switching of MPyAAm, we conducted 1H NMR analysis of MPyAAm ([MPyAAm]
0=0.1 mol L–1) in CD2Cl2 at 0°C
in the absence or presence of TFA (Figure 3). The signals of pyridyl and vinyl groups shifted significantly upon adding TFA, suggesting interaction between MPyAAm and TFA. In the pyridyl group resonances, small signals were observed on adding an equimolar amount of TFA. The small signals were also observed in the presence of 2-
and 3-fold amounts, but disappeared in the presence of 4- and 5-fold amounts of TFA. It was assumed that the small signals were assignable to the pyridyl groups in the s-cis O=C-N-Py conformer that is in equilibrium with the original s-trans O=C-N-Py conformer, and the equilibrium exchange rate was enhanced by excess TFA. The details are discussed later.
<Figure 3>
In the vinyl group resonances, signals assignable to the s-cis O=C-N-Py conformer were not clearly observed, probably because of overlap of the signals due to the two conformers. Protonation of the carbonyl group would result in a down-field shift of the signals of the vinylidene protons and an up-field shift of the signal of the methine proton.25 Indeed, the signals of the vinylidene protons moved gradually towards
lower magnetic field with an increase in the amount of added TFA. However, the methine proton signal moved slightly towards higher magnetic field upon adding an equimolar amount of TFA, then moved towards lower magnetic field upon adding excess TFA. The down-field shift in the presence of excess TFA suggests conformational switching of MPyAAm by protonation, because the methine proton should become free from the shielding effect of the pyridyl ring in the s-trans O=C-N-Py conformation when conformational switching takes place.26
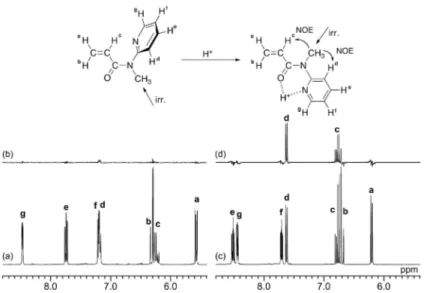
NOE difference spectroscopy was performed to confirm the conformational switching. Saturation of the N-methyl group led to significant NOE enhancement for
both the Hc in the vinyl group and the Hd proton at the 3-position in the pyridyl group in
the presence of TFA, although no NOE was observed in the absence of TFA (Figure 4). Furthermore, the NOE enhancement increased linearly with increase in the amount of added TFA, although 4-fold and 5-fold amounts of TFA exhibited similar NOE enhancements (Figure 5). It should be noted that this tendency was similar to the dependence of the m dyad content on the amount of the added TFA (cf. Figure 2).
<Figure 4> <Figure 5>
13C NMR spectra of MPyAAm ([MPyAAm]
0=0.1 mol L–1) in the absence or
presence of TFA were also measured in CD2Cl2 at 0°C. The 13C NMR signals of
MPyAAm shifted significantly on adding TFA as well as the 1H NMR signals. Figure 6
displays the changes in the chemical shifts of the acryl group and the pyridyl group on adding TFA.
<Figure 6>
By adding equimolar and 2-fold amounts of TFA, the signals of the vinylidene and carbonyl carbons exhibited down-field shifts. This result supports protonation of the carbonyl group. Furthermore, the signal of the carbon at the 4-position in the pyridyl group exhibited a down-field shift, and those at the 2- and 6-positions in the pyridyl
group exhibited up-field shifts. These results indicate that the pyridyl group was also protonated.27
By adding a 3-fold amount of TFA, the changes in the chemical shifts of all of the carbons were substantial. However, significant changes were not observed between 4-fold and 5-fold amounts of TFA. These results imply that the conformational switching of MPyAAm required at least a 3-fold amount of TFA.
Proposed Mechanism for Conformational Switching of MPyAAm with Protonation It has been proposed that N-aryl-N-methylamides favor the s-trans O=C-N-Ar conformation, because the s-cis O=C-N-Ar conformation is destabilized by the electronic repulsion between the carbonyl lone-pair electrons and the aryl π-electrons in the aryl ring twisted by steric hindrance between the N-methyl group and the aryl ring.15,28 In fact, the major conformation dramatically changed from the s-trans
O=C-N-Ar to the s-cis O=C-N-Ar upon addition of TFA in the case of N-[3,5-bis(dimethylamino)phenyl]-N-methylacetamide, probably because of the decrease in the electronic repulsion with protonation at the amino groups.28
In the present system, conformational switching could occur via the following mechanism (Scheme 1):
1. both the carbonyl and pyridyl groups of structure A are protonated to become structure C via structure B or B’, resulting in a decrease in the electronic repulsion between the carbonyl and pyridyl groups;
the positive charges prevents structure C from converting;
3. however, structure C is transformed into structure D, accompanied by abstraction of one of the protons in structure C, probably catalyzed by the conjugate base of the third acid, to form double protonations of both the carbonyl and pyridyl groups by one proton.
If the conjugate base of the third acid catalyzed the conformational switching, excess TFA should enhance the equilibrium exchange. This corresponds to the result that the 1H NMR signals of the s-trans and s-cis O=C-N-Py conformers were not
distinguished in the presence of 4-fold and 5-fold amounts of TFA (cf. Figure 3). Furthermore, it is explicable that MSA exhibited the isotactic-specificity-inducing effect to a lesser extent than TFA (cf. Table 1).
When conformational switching takes place via the structure B, the concentration of D can be expressed by the following equation:
[D]=K1BK2BK3[A][H+]
where K1B, K2B, and K3 are the equilibrium constants for the protonation of A to B, and
for B to C, and for conformational switching of C to D, respectively. Even when the conformational switching takes place via the structure B’, the concentration of D can be expressed by a similar equation:
[D]=K1BK2BK3[A][H+]
These equations indicate that the concentration of D is proportional to the concentrations of A and H+. This corresponds to linear increases of the m dyad content of the polymers obtained (cf. Figure 2) and of the NOE enhancement in 1H NMR of
MPyAAm (cf. Figure 5), with the amount of added TFA. Furthermore, the conformational switching of C to D accompanies release of one of two H+ in C. This means that increase in the concentration of H+ dose not simply result in acceleration of
the conformational switching, even though the conjugate base of the third acid catalyzed the conformational switching. Consequently, excess amount, such as 5-fold amount, of TFA was required to significantly induce the conformational switching.
CONCLUSION
MPyAAm was designed as a monomer that is conformationally switchable by protonation. The stereospecificity of radical polymerization of MPyAAm was changed from syndiotactic to isotactic by adding TFA in CH2Cl2 at low temperatures, as
expected. NMR analysis of mixtures of MPyAAm and TFA revealed that MPyAAm favored the s-cis O=C-N-Py conformation in the presence of excess TFA, and the s-trans O=C-N-Py conformation in the absence of TFA. Consequently, it is suggested that conformational switching of the monomer with protonation was responsible for the change in the stereospecificity of the radical polymerization of MPyAAm. Successful control of stereospecificity of radical polymerization by conformational switching of monomer would allow development of novel methodology for control of stereospecificity of radical polymerization.
REFERENCES AND NOTES
7180-7181.
2. Habaue, S.; Isobe, Y.; Okamoto, Y. Tetrahedron 2002, 58, 8205-8209. 3. Hirano, T.; Miki, H.; Seno, M.; Sato, T. Polymer 2005, 46, 3693-3699. 4. Hirano, T.; Ishizu, H.; Sato, T. Polymer 2008, 49, 438-445.
5. Hirano, T.; Okumura, Y.; Kitajima, H.; Seno, M.; Sato, T. J Polym Sci Part A: Polym Chem 2006, 44, 4450-4460.
6. Hirano, T.; Nakamura, K.; Kamikubo, T.; Ishii, S.; Tani, K.; Mori, T.; Sato, T. J Polym Sci Part A: Polym Chem 2008, 46, 4575-4583.
7. Hirano, T.; Kamikubo, T.; Okumura, Y.; Bando, Y.; Yamaoka, R.; Mori, T.; Ute, K. J Polym Sci Part A: Polym Chem 2009, 47, 2539-2550.
8. Hirano, T.; Yamaoka, R.; Miyazaki, T.; Ute, K. J Polym Sci Part A: Polym Chem 2010, 48, 5718-5726.
9. Liu, W.; Nakano, T.; Okamoto, Y. Polym J 2000, 32, 771-777.
10. Hirano, T.; Nasu, S.; Morikami, A.; Ute, K. J Polym Sci Part A: Polym Chem 2009, 47, 6534-6539.
11. Pedersen, B. F. Acta Chem Scand 1967, 21, 1415-1424.
12. Itai, A.; Toriumi, Y.; Tomioka, N.; Kagechika, H.; Azumaya, I.; Shudo, K. Tetrahedron Lett 1989, 30, 6177-6180.
13. Yamaguchi, K.; Matsumura, G.; Kagechika, H.; Azumaya, I.; Ito, Y.; Itai, A.; Shudo, K. J Am Chem Soc 1991, 113, 5474-5475.
14. Itai, A.; Toriumi, Y.; Saito, S.; Kagechika, H.; Shudo, K. J Am Chem Soc 1992, 114, 10649-10650.
15. Saito, S.; Toriumi, Y.; Tomioka, N.; Itai, A. J Org Chem 1995, 60, 4715-4720. 16. Tanaka, H.; Niwa, M. Polymer 2008, 49, 3693-3701.
17. Okamoto, I.; Nabeta, M.; Minami, T.; Nakashima, A.; Morita, N.; Takeya, T.; Masu, H.; Azumaya, I.; Tamura, O. Tetrahedron Lett 2007, 48, 573-577.
18. Okamoto, I.; Nabeta, M.; Hayakawa, Y.; Morita, N.; Takeya, T.; Masu, H.; Azumaya, I.; Tamura, O. J Am Chem Soc 2007, 129, 1892-1893.
19. Buback, M.; Kurz, C. H. Macromol Chem Phys 1998, 199, 2301-2310. 20. Beuermann, S.; Nelke, D. Macromol Chem Phys 2003, 204, 460-470. 21. Beuermann, S. Macromolecules 2004, 37, 1037-1041.
22. Lee, T. Y.; Roper, T. M.; Jönsson, E. S.; Guymon, C. A.; Hoyle, C. E. Macromolecules 2004, 37, 3659-3665.
23. Bordwell, F. G. Acc Chem Res 1988, 21, 456-463.
24. Hirano, T.; Masuda, S.; Nasu, S.; Ute, K.; Sato, T. J Polym Sci Part A: Polym Chem 2009, 47, 1192-1203.
25. The signals of vinylidene protons and methine proton of DMAAm, as measured in toluene-d8 at 0°C, exhibited down-field and up-field shifts by mixing with diethyl
L-tartrates which form a complex with DMAAm though double C=O•••H-O hydrogen bonding in toluene at low temperatures.
26. The signal of the methine proton of DMAAm was observed at lower magnetic field than those of MPyAAm and MPhAAm, as measured in CDCl3 at 25°C.
27. In the 13C NMR spectra of pyridine in CDCl
3 25°C, on adding TFA the signal of
at the 2- and 6-positions shifted up-field.
28. Yamasaki, R.; Tanatani, A.; Azumaya, I.; Saito, S.; Yamaguchi, K.; Kagechika, H. Org Lett 2003, 5, 1265-1267.
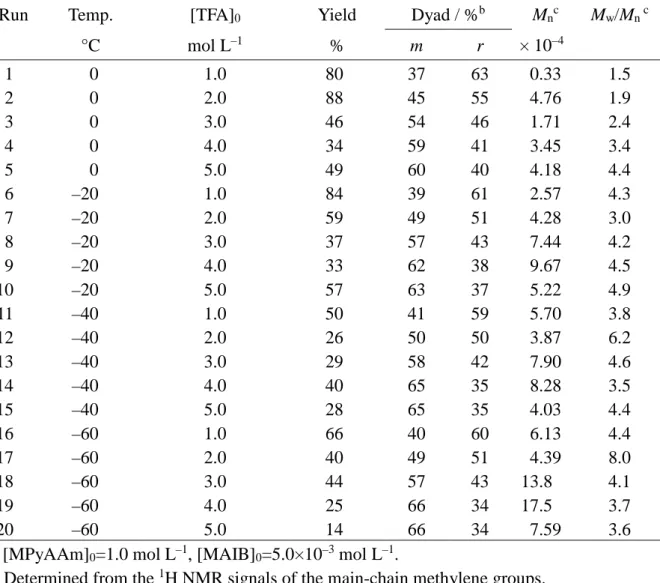
Table 1. Radical polymerization of MPyAAm in CH3CN at low temperatures for 16 h in
the presence of TFA or MSA.a
Run Temp. Additive [Additive]0 Yield Dyad / %b Mnc Mw/Mn c
°C mol L–1 % m r × 10–4 1 2 3 4 5 6 7 8 9 10 11 12 0 0 0 –40 –40 –40 0 0 0 –40 –40 –40 TFA TFA TFA TFA TFA TFA MSA MSA MSA MSA MSA MSA 1.0 3.0 5.0 1.0 3.0 5.0 1.0 3.0 5.0 1.0 3.0 5.0 48 55 50 60 54 30 >99 58 >99 29 49 51 38 44 44 44 47 47 42 49 49 45 48 47 62 56 56 56 53 53 58 51 51 55 52 53 0.63 2.05 4.36 2.59 3.75 5.20 1.26 1.82 3.08 ndd 0.73 0.93 1.8 3.1 3.1 2.6 3.2 4.4 3.2 4.7 5.1 ndd 2.9 2.3 a. [MPyAAm]0=1.0 mol L–1, [MAIB]0=5.0×10–3 mol L–1.
b. Determined from the 1H NMR signals of the main-chain methylene groups. c. Determined by SEC (polystyrene standards) for poly(MPyAAm) hydrochloride. d. Not determined.
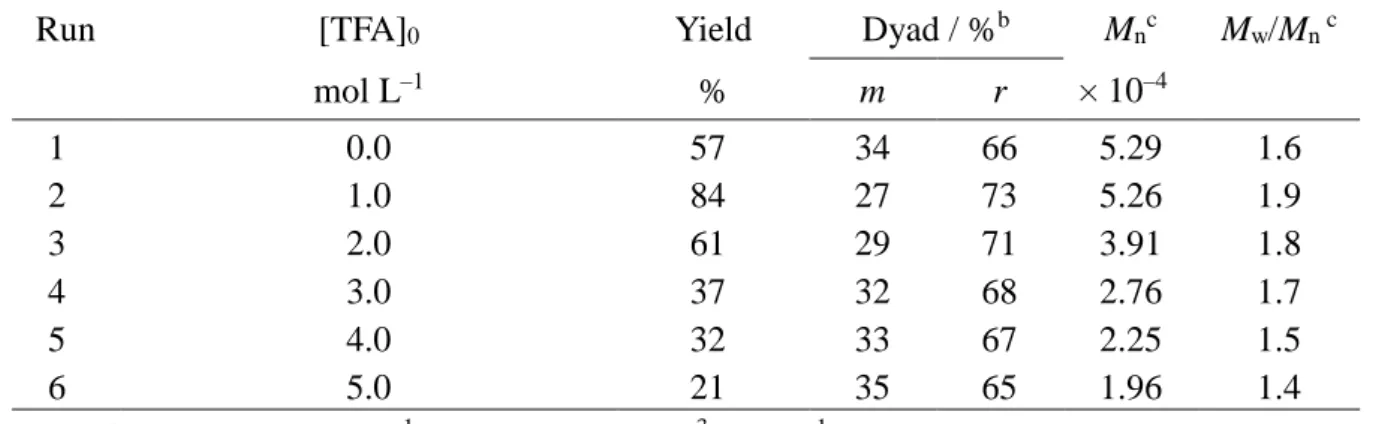
Table 2. Radical polymerization of MPyAAm in CH2Cl2 at low temperatures for 16 h in
the presence of TFAa.
Run Temp. [TFA]0 Yield Dyad / %b Mnc Mw/Mn c
°C mol L–1 % m r × 10–4 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 0 0 0 0 0 –20 –20 –20 –20 –20 –40 –40 –40 –40 –40 –60 –60 –60 –60 –60 1.0 2.0 3.0 4.0 5.0 1.0 2.0 3.0 4.0 5.0 1.0 2.0 3.0 4.0 5.0 1.0 2.0 3.0 4.0 5.0 80 88 46 34 49 84 59 37 33 57 50 26 29 40 28 66 40 44 25 14 37 45 54 59 60 39 49 57 62 63 41 50 58 65 65 40 49 57 66 66 63 55 46 41 40 61 51 43 38 37 59 50 42 35 35 60 51 43 34 34 0.33 4.76 1.71 3.45 4.18 2.57 4.28 7.44 9.67 5.22 5.70 3.87 7.90 8.28 4.03 6.13 4.39 13.8 17.5 7.59 1.5 1.9 2.4 3.4 4.4 4.3 3.0 4.2 4.5 4.9 3.8 6.2 4.6 3.5 4.4 4.4 8.0 4.1 3.7 3.6 a. [MPyAAm]0=1.0 mol L–1, [MAIB]0=5.0×10–3 mol L–1.
b. Determined from the 1H NMR signals of the main-chain methylene groups. c. Determined by SEC (polystyrene standards) for poly(MPyAAm) hydrochloride.
Table 3. Radical polymerization of MPhAAm in CH2Cl2 at 0°C for 16 h in the presence
or absence of TFAa.
Run [TFA]0 Yield Dyad / %b Mnc Mw/Mn c
mol L–1 % m r × 10–4 1 2 3 4 5 6 0.0 1.0 2.0 3.0 4.0 5.0 57 84 61 37 32 21 34 27 29 32 33 35 66 73 71 68 67 65 5.29 5.26 3.91 2.76 2.25 1.96 1.6 1.9 1.8 1.7 1.5 1.4 a. [MPhAAm]0=1.0 mol L–1, [MAIB]0=5.0×10–3 mol L–1.
b. Determined from the 1H NMR signals of the main-chain methylene groups. c. Determined by SEC (polystyrene standards).
Figure 1. 1H NMR spectra of the methylene groups in the main chain of poly(MPyAAm)s prepared in CH2Cl2 at 0°C in the presence of (a) 1 equiv., (b) 2 equiv.,
Figure 2. Relationship between the [TFA]0/[MPyAAm]0 ratio and the m dyad contents
of poly(MPyAAm)s prepared at –60°C to 0°C. The inset shows 1H NMR spectrum of the methylene group in the main chain of poly(MPyAAm) prepared in CH2Cl2 at –60°C in the
Figure 3. Expanded 1H NMR signals of the vinyl group and the pyridyl group of MPyAAm (0.1 mol L–1) in CD2Cl2 at 0°C. ([TFA]0: (a) 0.0 mol L–1, (b) 0.1 mol L–1, (c)
Figure 4. 1H and NOE difference spectra of MPyAAm in CD2Cl2 at 0°C in the (a, b)
absence or (c, d) presence of a 5-fold amount of TFA, saturating the signal of the N-methyl group.
Figure 5. Relationship between the [TFA]0/[MPyAAm]0 ratio and the NOE
Figure 6. Chemical shift changes in 13C NMR spectra of (a) acryloyl group and (b) pyridyl group in MPyAAm upon addition of TFA.
Scheme 1. Proposed mechanism for the conformational switching of MPyAAm with protonation.

![Figure 2. Relationship between the [TFA] 0 /[MPyAAm] 0 ratio and the m dyad contents of poly(MPyAAm)s prepared at –60°C to 0°C](https://thumb-ap.123doks.com/thumbv2/123deta/6781842.1163412/23.892.240.666.151.647/figure-relationship-tfa-mpyaam-ratio-contents-mpyaam-prepared.webp)
![Figure 5. Relationship between the [TFA] 0 /[MPyAAm] 0 ratio and the NOE enhancements observed for the H c in the vinyl group and the H d in the pyridyl group](https://thumb-ap.123doks.com/thumbv2/123deta/6781842.1163412/26.892.265.642.203.646/figure-relationship-mpyaam-ratio-enhancements-observed-vinyl-pyridyl.webp)