Studies on the molecular mechanisms of skin
pigmentation
著者
Marubashi Sojiro
学位授与機関
Tohoku University
学位授与番号
11301甲第19369号
URL
http://hdl.handle.net/10097/00127815
博士論文
Studies on the molecular mechanisms of skin pigmentation
(皮膚暗色化に関する分子メカニズムの研究)
令和元年度 東北大学大学院 生命科学研究科 生命機能科学専攻 丸橋 総史郎Contents
Overview ………...……… 3
Abbreviations ……… 5
Chapter 1: A novel Varp-binding protein, RACK1, regulates dendrite outgrowth through stabilization of Varp protein in melanocytes Abstract ………...……… 7
Introduction ……..………..……… 8
Materials and Methods ….……….………..……….…… 11
Results ……….………. 17
Discussion ……… 21
References ……… 24
Figures and Figure Legends ………..……… 29
Chapter 2: Rab7B/42 is functionally involved in protein degradation on melanosomes in keratinocytes Abstract ……… 43
Introduction ……..……… 44
Materials and Methods ….……….……….…… 46
Results ……….………. 52
Discussion ……… 57
References ……… 59
Figures and Figure Legends ………...………..……… 64
Overview
Melanosomes are specialized organelles where melanin pigment is synthesized by melanogenic enzymes in mammalian skin melanocytes. Mature darkly pigmented melanosomes are transported to the periphery of the melanocytes along the cytoskeleton, and they are ultimately transferred to surrounding keratinocytes through the melanocyte dendrites, specialized cell structures that can contact surrounding keratinocytes. During the past few decades, a number of proteins involved in the biogenesis and transport of melanosomes and their functions in melanocytes have gradually been elucidated, but the regulatory mechanism of those proteins are poorly understood. Moreover, the proteins that are related to subsequent processes such as accumulation and decomposition of the incorporated melanosomes in keratinocytes are poorly identified. In this thesis, I report two mechanisms involving mammalian skin pigmentation: “RACK1 regulates dendrite outgrowth in melanocytes (Chapter 1)” and “Rab7B/42 promotes protein degradation on melanosomes in keratinocytes (Chapter 2)”.
In Chapter 1, I focused on the regulatory mechanism of Varp (VPS9-ankyrin repeat protein) protein expression. Previous studies from our groups have shown that Varp is a pleiotropic regulator of melanogenesis; it promotes dendrite formation as well as melanogenic enzyme transport to melanosomes. Furthermore, the Varp protein level has been shown to be negatively regulated by proteasomal degradation through interaction with the small GTPase Rab40C. However, the molecular mechanisms by which Varp escapes from Rab40C and retains its own expression level remain completely unknown. In this chapter, I screened for a Varp binding protein and succeeded in identifying RACK1 (receptor of activated protein kinase
C 1) as a novel Varp binding partner. I found that knockdown of endogenous RACK1 in melanocytes caused dramatic reduction of the Varp protein level and inhibition of dendrite outgrowth. Furthermore, competitive co-immunoprecipitation assay showed that RACK1 inhibits the interaction between Varp and Rab40C. These findings indicated that RACK1 competes with Rab40C for binding to Varp and regulates dendrite outgrowth through stabilization of Varp in melanocytes.
In Chapter 2, I focused on the molecular basis of melanosome uptake and decomposition in keratinocytes. Although several proteins such as LAMP1 (lysosomal-associated membrane protein 1) have been observed in melanosome-containing compartments in keratinocytes, no specific marker protein for such compartments has been identified, and no appropriate assay to assess the degradation of proteins on melanosomes in keratinocytes has been established. In the second chapter, I performed a comprehensive localization screening for mammalian Rab family small GTPases (Rab1~45) and succeeded in identifying 11 Rabs that were enriched around melanosomes that had been incorporated into keratinocytes. I also developed a new assay to quantitatively assess the degradation of proteins on such melanosomes in keratinocytes and found that depletion of Rab7B (also identified as Rab42) in keratinocytes resulted in the strongest inhibition of protein degradation on incorporated melanosomes. These results indicated that Rab7B/42 is recruited to melanosome-containing compartments and that it promotes protein degradation on melanosomes in keratinocytes.
Abbreviations
ANKR: ankyrin repeatEEA1: early endosomal antigen 1
EGFP: enhanced green fluorescent protein Fsk: forskolin
HRP: horseradish peroxidase KD: knockdown
KO: knockout
LAMP1: lysosomal-associated membrane protein 1 LBPA: lysobisphosphatidic acid
LysoT: LysoTracker Red mCherry: monomeric Cherry mStr: monomeric Strawberry
M-INK: Melanocore-INteracting Kif1c-tail PBS: phosphate-buffered saline
RACK1: receptor of activated protein kinase C 1 RFP: red fluorescent protein
RT: reverse transcription siRNA: small interfering RNA SC: synthetic complete
TPA: 12-O-tetradecanoylphorbol 13-acetate Tyrp1: tyrosinase-related protein 1
Chapter 1
A novel Varp-binding protein, RACK1, regulates dendrite outgrowth through
stabilization of Varp protein in melanocytes
Abstract
Varp in melanocytes is thought to function as a key player in pigmentation of mammals. Varp regulates two different melanocyte functions, i.e., transport of melanogenic enzymes to melanosomes by functioning as a Rab32/38 effector, and promotion of dendrite outgrowth by functioning as a Rab21-guanine nucleotide exchange factor. The Varp protein level has recently been shown to be negatively regulated by proteasomal degradation through interaction of the ankyrin repeat 2 (ANKR2) domain of Varp with Rab40C. However, the molecular mechanisms by which Varp escapes from Rab40C and retains its own expression level remain completely unknown. In this thesis, I identified RACK1 as a novel Varp-ANKR2-binding partner and investigated its involvement in Varp stabilization in mouse melanocytes. The results showed that knockdown of endogenous RACK1 in melanocytes caused dramatic reduction of the Varp protein level and inhibition of dendrite outgrowth, and intriguingly, overexpression of RACK1 inhibited the interaction between Varp and Rab40C and counteracted the negative effect of Rab40C on dendrite outgrowth. These findings indicated that RACK1 competes with Rab40C for binding to the ANKR2 domain of Varp and regulates dendrite outgrowth through stabilization of Varp in mouse melanocytes.
Introduction
Melanocytes are unique cells that play an important role in producing pigmented organelles, called melanosomes, that protect cells against ultraviolet injury (Raposo and Marks, 2007). Melanocytes originate from the embryonic neural crest cells of mammals, and the embryonic neural crest cells migrate and differentiate into melanocytes in the basal layer of the epidermis and in the hair bulbs (Cichorek et al., 2013). Skin and hair become pigmented as a result of a process that consists of several steps. In the first step, mature melanosomes are produced around the nucleus (i.e., melanosome biogenesis step). In the next step, the mature melanosomes are transported (i.e., melanosome transport step) along the two cytoskeletal components, microtubules and actin filaments, and then transferred to surrounding keratinocytes through the melanocyte dendrites (i.e., melanosome transfer step). The melanocyte dendrites are specialized cell structures that form and contact surrounding keratinocytes and hair matrix cells (i.e., dendrite outgrowth step) in response to growth factors and ultraviolet irradiation (Ohbayashi and Fukuda, 2012; Wu and Hammer, 2014). Each of these steps is thought to be crucial to pigmentation, because genetic defects in one of these steps have been reported to cause albinism, a group of hereditary diseases that are characterized by hypopigmentation of the hair and skin, e.g., Griscelli syndrome, Hermansky-Pudlak syndrome, and Chédiak-Higashi syndrome (Tomita and Suzuki, 2004; Di Pietro and Dell’Angelica, 2005; Van Gele et al., 2009). Genetic analysis of these diseases and their corresponding coat color mutant mice in the past few decades have revealed their causative genes, and further characterization of the gene products has enabled identification of a variety of their binding proteins and regulators that participate in skin and hair
pigmentation, e.g., BLOC (biogenesis of lysosome-related organelles complex) (Wei and Li, 2013) and small GTPase Rab38 (Loftus et al., 2002).
Varp is one such protein that functions as an effector molecule for the small GTPase Rab38 (Tamura et al., 2009), a deficiency of which causes the diluted coat color of chocolate mice (Loftus et al., 2002), one of the mouse models of Hermansky-Pudlak syndrome (Wei and Li, 2013). Varp is a pleiotropic regulator of melanogenesis, because it promotes dendrite formation through activation of Rab21 via its N-terminal VPS9 domain (Zhang et al., 2006; Ohbayashi et al., 2012b), and melanogenic enzyme transport to melanosomes via its ankyrin repeat 1 (ANKR1) domain, which binds Rab32/38 (Wang et al., 2008; Tamura et al., 2009; Tamura et al., 2011). In addition, binding of VAMP7 to a VAMP7-interaction domain (VID) of Varp (Burgo et al., 2009; Schäfer et al., 2012) is required for both dendrite outgrowth and melanogenic enzyme transport (Tamura et al., 2011). The results of recent proteomic analyses have revealed the presence of additional Varp binding partners, including retromer, GolginA4, and Kif5A, in non-melanocytic cells (Burgo et al., 2012; Hesketh et al., 2014; McGough et al., 2014), but their involvement in melanogenesis is poorly understood. More recently, it has been reported that Rab40C binds to a C-terminal ANKR2 domain of Varp and that the binding promotes proteasomal degradation of Varp through recruiting a Cullin-type ubiquitin ligase complex (Yatsu et al., 2015). However, the molecular mechanism by which Varp escapes from Rab40C-mediated degradation remains completely unknown, and the presence of an additional as yet unknown Varp binding partner(s) that regulates the interaction between Rab40C and the ANKR2 domain of Varp in melanocytes is assumed.
In this chapter, I screened for a novel Varp ANKR2-domain-binding protein by performing yeast-two hybrid assays and succeeded in identifying RACK1 (also known as Gnb2l1
in the NCBI database) as a candidate. RACK1 was originally described as an anchoring protein for activated PKCβΙΙ (Ron et al., 1994), and it has subsequently been shown to function as a scaffold protein that is involved in a variety of cellular events, including translation, apoptosis, and migration (Serrels et al., 2011; Gandin et al., 2013; Li and Xie, 2014). The results of biochemical assays showed that the interaction between RACK1 and Varp was increased by forskolin, which increased the intracellular cAMP concentration and induced dendrite outgrowth of melanocytes (Ohbayashi et al., 2012b), and that RACK1 had the ability to reduce Rab40C binding to the ANKR2 domain of Varp by competitively binding to it. Moreover, knockdown of endogenous RACK1 in melanocytes in this study caused a dramatic reduction in the amount of Varp protein and in forskolin-induced dendrite outgrowth. Based on my findings, I discuss a novel role of RACK1 in protecting Varp protein from Rab40C during dendrite outgrowth of melanocytes and its relation to albinism.
Materials and Methods
MaterialsAnti-Varp guinea pig polyclonal antibody and anti-tyrosinase rabbit polyclonal antibody were prepared as described previously (Beaumont et al., 2011; Yatsu et al., 2015). The following antibodies used in this study were obtained commercially: anti-RACK1 mouse monoclonal antibody (Santa Cruz Biotechnology, Dallas, TX), anti-β-actin mouse monoclonal antibody
(Applied Biological Materials, Richmond, BC, Canada), HRP (horseradish
peroxidase)-conjugated anti-FLAG tag (M2) mouse monoclonal antibody, anti-FLAG tag antibody-conjugated agarose (Sigma-Aldrich, St. Louis, MO), HRP-conjugated anti-T7 tag mouse monoclonal antibody (Merck Millipore, Billerica, MA), and HRP-conjugated anti-HA tag (3F10) rat monoclonal antibody (Roche Applied Science, Penzberg, Germany). Forskolin and TPA were from Sigma-Aldrich. The proteasome inhibitor MG132 and N-ethylmaleimide were obtained from Peptide Institute (Osaka, Japan) and FUJIFILM Wako Pure Chemical Industries (Osaka, Japan), respectively.
Plasmid construction
Plasmids encoding mouse Varp (N+VPS9, ANKR1, and ANKR2), Rab40C, and EGFP were prepared as described previously (Tamura et al., 2009; Yatsu et al., 2015). The cDNA of Varp-ANKR2 (amino acid residues 730–1048) (Yatsu et al., 2015) was subcloned into the pGBKT7 vector (Takara Bio Inc., Shiga, Japan) for yeast two-hybrid screening. The cDNA of mouse RACK1 was amplified from Marathon-Ready mouse brain and testis cDNAs (Takara Bio Inc.) by performing PCR with the following pair of oligonucleotides containing a BamHI linker
(underlined) or a stop codon (boldface): forward primer,
5’-GGATCCATGACCGAGCAGATGACCCT-3’ and reverse primer,
5’-TTAGCGGGTACCAATAGTTA-3’. The RACK1 cDNAs obtained were subcloned into the pEF-T7 tag vector (Fukuda et al., 1999), pEF-HA tag vector (Fukuda, 2002), pmStr-C1 vector (Ohbayashi et al., 2012a), and pGBD-C1 vector (James et al., 1996). A RACK1ΔN mutant (deletion of amino acid residues 1–203 of mouse RACK1) was prepared by removing the BamHI insert of the pGBD-C1-RACK1 vector (an intrinsic BamHI site in the RACK1 cDNA and BamHI site in the multi-cloning site of the vector) and by self-ligating the vector. Small interfering RNAs (siRNAs) against mouse RACK1 (target site 1: 5’-GGATGAGAGTCATTCAGAA-3’ and
target site 2; 5’-GACCAACTATGGCATACCA-3’), mouse Varp (target site:
5’-GGAAACAGGAUUAAGCUAG-3’) and mouse Rab40C (target site:
5’-CTGCATGACCTTCTTTGAA-3’) were chemically synthesized by Nippon Gene Co., Ltd. (Toyama, Japan). Knockdown efficiency of the Varp and Rab40c siRNA has already been shown in previous study (Tamura et al., 2009; Yatsu et al., 2015).
Yeast two-hybrid assays
Yeast two-hybrid screening was performed by using pGBKT7-Varp-ANKR2 as bait and mouse testis and mouse 11-day embryo mixed cDNA libraries (Takara Bio Inc.) as prey according to the manufacturer’s instructions. Yeast two-hybrid assays were performed by using pGBD-C1-RACK1ΔN and pAct2-Varp-N+VPS9, pAct2-Varp-ANKR1, or pAct2-Varp-ANKR2 as described previously (Tamura et al., 2009). The yeast strain (pJ69-4A), SC-LW medium (synthetic complete medium lacking leucine and tryptophan), selection medium SC-AHLW
(synthetic complete medium lacking adenine, histidine, leucine, and tryptophan), culture conditions, and transformation protocol used are described elsewhere (James et al., 1996; Kobayashi et al., 2015). Yeast cells containing both pGBD-C1-RACK1ΔN and each of the pAct2 plasmids expressing a Varp mutant were each streaked on both SC-LW medium and SC-AHLW medium and incubated at 30˚C for 1 day and 1 week, respectively.
RT (reverse transcription)-PCR analysis
The total RNA of melan-a cells transfected with RACK1 siRNA (st1 or st2) or control siRNA was prepared with TRI-reagent (Sigma-Aldrich), and reverse transcription was performed by using ReverTra Ace® (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. Real-time quantitative PCR was performed with SYBR Green and Light Cycler 2.0 (Roche Applied Science) according to the manufacturer’s instructions. The following pairs of oligonucleotides were used for amplification: for RACK1, forward primer, 5’-GTACACGGTCCAGGATGAG-3’ and reverse primer, 5’-GGCCAATGTGGTTGGTCTTTA-3’; for Varp, forward primer,
5’-ACATCCTGAACAAGAGGCAGTA-3’ and reverse primer,
5’-CCTGATCTCAACAGTCACGTA-3’; for tyrosinase, forward primer,
5’-GGGATGAGAACTTCACTGTTCCATA-3’ and reverse primer,
TGATCTGCTACAAATGATCTGCC-3’; and for GAPDH, forward primer, 5’-CATGGCCTTCCGTGTTCCTA-3’ and reverse primer, 5’- GAGTTGCTGTTGAAGTCGC-3’. qPCR data were normalized to GAPDH and expressed as means and SEM of four independent experiments.
Forskolin-induced dendrite formation of melanocytes
The black-mouse-derived immortal melanocyte cell line melan-a (generous gift of Dorothy C. Bennett) was cultured on 12-mm diameter coverslips as described previously (Bennett et al., 1987; Kuroda et al., 2003). Plasmids and siRNAs were transfected into melan-a cells by using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. One day after transfection the cells were treated with 20 µM forskolin or DMSO alone (control), and 20 hours later they were fixed with 4% paraformaldehyde and examined for fluorescence with a confocal laser-scanning fluorescence microscope (Fluoview FV1000-D; Olympus, Tokyo, Japan). The images of the EGFP-expressing cells were captured at random with the confocal microscope, and the total length of their dendrites was measured with ImageJ software (version1.48u4; NIH, Bethesda, MD) as described previously (Ohbayashi et al., 2012b). In brief, the length of each dendrite of a cell was measured from the edge of the cell body (cell body–dendrite boundary was determined to be a cross point by extrapolating the cell membrane) to the tip of dendrite, and total dendrite length means the sum of the lengths of all of the dendrites of the cell. Each experiment was performed independently at least three times, and the numbers of cells analyzed in each experiment are stated in the figure legends. The statistical analyses were performed by using Dunnett’s test, Tukey’s test, or Student’s unpaired t-test, and P values <0.05 were considered statistically significant (*, P<0.05; **, P<0.01).
Immunoblotting
siRNAs were transfected into melan-a cells by using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. At 48 hours after transfection the cells
mM NaCl, 1 mM MgCl2, and 1% Triton X-100 supplemented with complete EDTA-free protease inhibitor cocktail [Roche Applied Science]). In Figure 2d, at 6 hours after transfection the cells were treated with 1 nM proteasome inhibitor MG132 or DMSO and then were cultured for 48 hours. The cells were washed with ice-cold PBS and lysed with 50 mM HEPES-KOH pH 7.2, 150 mM NaCl, 1 mM MgCl2, 0.1% (w/v) sodium deoxycholate, 1% (w/v) SDS, and 10 mM N-ethylmaleimide supplemented with complete EDTA-free protease inhibitor cocktail. The lysates were subjected to 7.5% (or 8.8%) SDS-PAGE and transferred to a PVDF membrane (Merck Millipore) by electroblotting. The blots were blocked with 1% skim milk or 1% BSA in PBS containing 0.1% Tween-20 and incubated at room temperature with primary antibodies for 1 hour and then with appropriate HRP-conjugated secondary antibodies for 1 hour. Immunoreactive bands were detected by enhanced chemiluminescence (ECL, GE Healthcare Ltd., Little Chalfont, UK). The intensity of the immunoreactive bands was measured with ImageJ software. The positions of the molecular mass markers (in kDa) are shown on the left of each figure.
Co-immunoprecipitation assays
COS-7 cells (4 × 105 cells/60-mm dish) were co-transfected with plasmids (indicated in Figures 1 and 4) by using Lipofectamine LTX Plus (Thermo Fisher Scientific) according to the manufacturer’s instructions. At 36 hours after transfection the cells were lysed with the lysis buffer. Co-immunoprecipitation assays with anti-FLAG tag antibody-conjugated agarose beads (Sigma-Aldrich) were performed as described previously (Fukuda et al., 1999; Fukuda and Kanno, 2005), and proteins bound to the beads were analyzed by immunoblotting as described above. Endogenous interaction between Varp and RACK1 in melan-a cells that had been
treated with 20 µM forskolin for 3 hours was also investigated by co-immunoprecipitation assays with anti-RACK1 antibody as described previously (Fukuda and Kanno, 2005).
Melanin assay and tyrosinase staining
The melanin content was assayed as described previously (Tamura et al., 2009). In brief, melan-a cells that had been transfected with RACK1 siRNAs were cultured for 4 days and their melanin content normalized to protein content was measured as optical density at 490 nm. Tyrosinase staining was also performed as described previously (Yatsu et al., 2015).
RESULTS
Identification of RACK1 as a novel Varp-binding protein
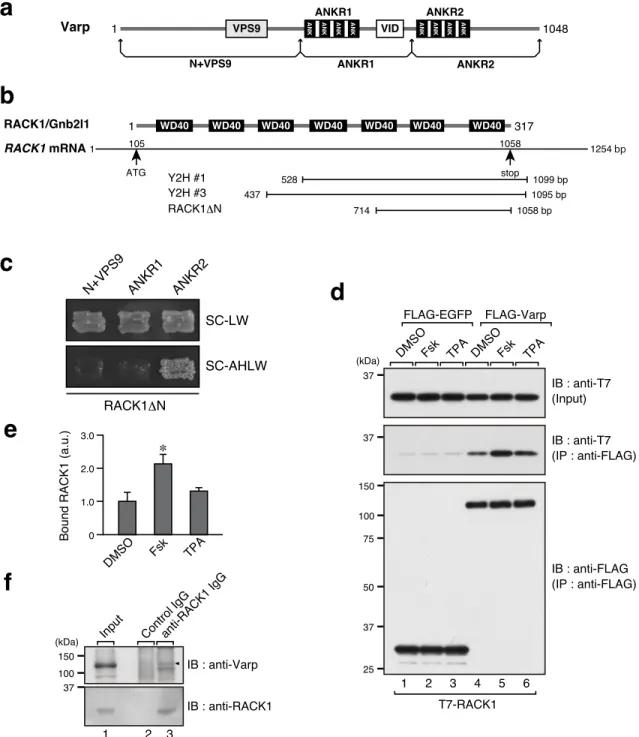
To identify novel Varp ANKR2-domain-binding partners, I performed yeast two-hybrid screening with the Varp ANKR2 domain used as bait (Figure 1a). I screened approximately 1.8 × 106 colonies of a mouse cDNA library and obtained 40 positive clones. I especially focused on the candidate protein, RACK1, because three independent prey clones were obtained for RACK1 (Figure 1b; clone #2 was not depicted because of its incomplete sequence), whereas only one or two positive clones were obtained for other candidate proteins. Because the two completely sequenced (#1 and #3) of the three RACK1 clones contained part of the C-terminal sequence of RACK1, I prepared a RACK1ΔN mutant (amino acids 204–317) and performed yeast two-hybrid assays to investigate its binding activity in relation to three Varp truncated mutants (Figure 1a). As expected, the RACK1ΔN construct specifically interacted with the ANKR2 domain and did not interact at all with the N+VPS9 domain or the ANKR1 domain (Figure 1c). In addition, I confirmed the interaction between full-length Varp and RACK1 in mammalian cultured cells by performing co-immunoprecipitation assays (Figure 1d, lane 4 in the middle panel). I also confirmed the endogenous interaction between Varp and RACK1 by using specific antibodies (Figure 1f). Because RACK1 has previously been shown to be involved in several signaling pathways, including a cAMP pathway and a PKC pathway (Adams et al., 2011), I treated cells with a PKC activator, 12-O-tetradecanoylphorbol 13-acetate (TPA), and a PKA activator, forskolin. Intriguingly, the interaction between Varp and RACK1 was significantly increased by forskolin, whereas TPA had no effect at all (Figure 1d, lanes 5 and 6 in the middle panel, and e).
Knockdown of RACK1 in melanocytes induces Varp degradation and inhibition of dendrite outgrowth
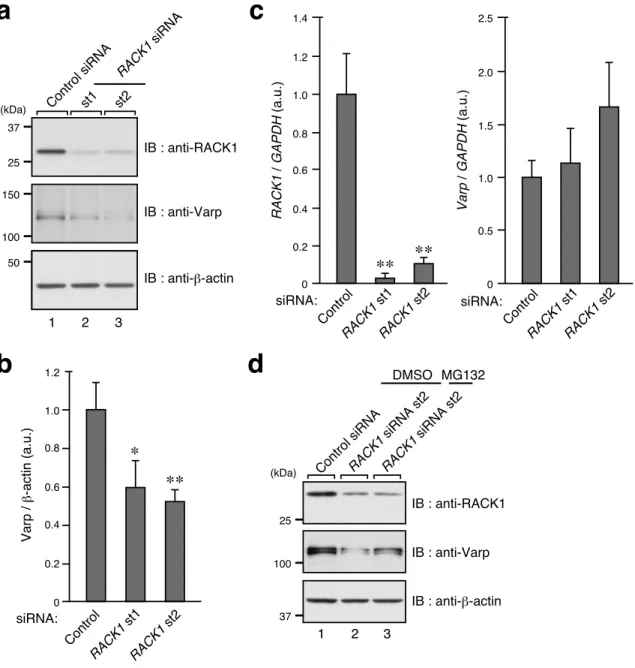
The finding that the Varp–RACK1 interaction was increased by forskolin led me to investigate the involvement of RACK1 in the forskolin-induced dendrite outgrowth of melanocytes (Ohbayashi et al., 2012b), because melanocyte dendrite outgrowth is the only forskolin-dependent phenomenon in which Varp has been reported to be involved in the literature. First, I investigated whether RACK1 is endogenously expressed in melanocytes. Since the results of immunoblotting and RT-PCR analysis indicated that RACK1 is actually expressed in mouse melan-a cells (Figure 2a, lane 1 in the top panel, and c, left graph), I proceeded to investigate whether RACK1 is involved in dendrite outgrowth of melanocytes. To do so, I prepared two independent sites of RACK1 siRNAs, both of which efficiently knocked down endogenous RACK1 protein (Figure 2a, top panel) as well as RACK1 mRNA in the melanocytes (Figure 2c, left graph), and the results showed that the knockdown of RACK1 strongly inhibited forskolin-induced dendrite outgrowth (Figure 3a and b), the same as Varp knockdown did (Ohbayashi et al., 2012b). Furthermore, knockdown of RACK1 also resulted in a significant decrease in the number of dendrites/cell (Control siRNA, 2.22 ± 0.11; RACK1 siRNA st1, 1.62 ± 0.09; and RACK1 siRNA st2, 1.27 ± 0.10 (mean ± SEM); P<0.01). To my surprise, there was a concomitant decrease in the amount of Varp protein in the RACK1-knockdown (KD) melanocytes (Figure 2a, middle panel, and b), and yet RACK1 knockdown had no significant effect on the level of Varp mRNA expression (Figure 2c, right graph). The lower level of Varp protein in the RACK1-KD cells is likely to have been attributable to proteasomal degradation,
protein level (Figure 2d, middle panel). These results indicated that RACK1 is most likely involved in dendrite outgrowth of forskolin-stimulated melanocytes through regulation of the Varp protein expression level.
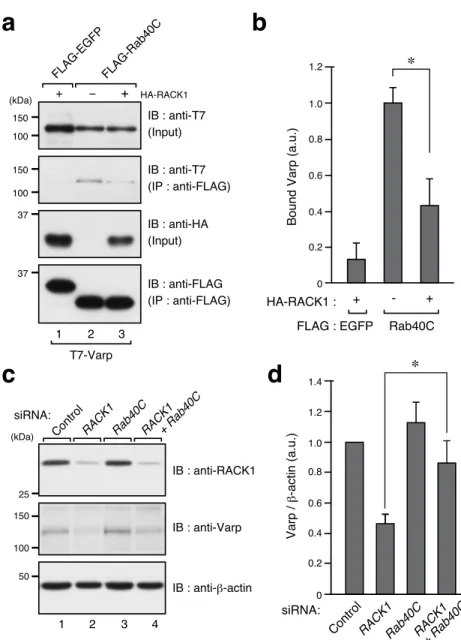
RACK1 inhibits the interaction between Varp and Rab40C
Because the Varp protein expression level is negatively controlled by Rab40C through proteasomal degradation and because Rab40C interacts with the ANKR2 domain of Varp (Yatsu et al., 2015), the same as RACK1 does (Figure 1c), I hypothesized that RACK1 protects Varp protein from proteasomal degradation by inhibiting the interaction between Rab40C and Varp. To test my hypothesis, I expressed T7-tagged Varp and FLAG-tagged Rab40C (or EGFP as a control) with or without HA-tagged RACK1 in COS-7 cells and performed co-immunoprecipitation assays (Figure 4a). The results showed that expression of HA-RACK1 dramatically decreased the amount of Varp that co-immunoprecipitated with Rab40C (Figure 4a, lanes 2 and 3 in the second panel, and b), indicating that RACK1 effectively inhibits the interaction between Varp and Rab40C in vitro. Consistent with this result, the effect of the RACK1 knockdown on the Varp protein level (i.e., decreased Varp protein level) in melanocytes was clearly attenuated by simultaneous knockdown of Rab40C (Figure 4c, lanes 2 and 4 in the middle panel, and d).
Finally, I investigated whether RACK1 antagonizes the effect of Rab40C on the Varp protein expression level during dendrite outgrowth. To do so, I expressed RACK1 with or without Rab40C in forskolin-stimulated melanocytes and evaluated its effect on dendrite outgrowth. Expression of mStr-RACK1 alone (+ EGFP as mock control) in
forskolin-stimulated melanocytes had no effect on dendrite outgrowth (Figure 5a, second row, and b), whereas expression of EGFP-Rab40C alone (+ mStr as mock control) significantly inhibited forskolin-induced dendrite outgrowth (Figure 5a, third row, and b). By contrast, simultaneous expression of EGFP-Rab40C and mStr-RACK1 in forskolin-stimulated melanocytes completely restored dendrite outgrowth to the mock control (EGFP/mStr) level (Figure 5a, bottom row, and b), and the rescue effect of RACK1 on dendrite outgrowth was canceled when endogenous Varp was knocked down in EGFP-Rab40C+mStr-RACK1-expressing cells (Figure 6). A similar tendency was observed in regard to the numbers of dendrites of the forskolin-stimulated cells: expression of EGFP-Rab40C alone slightly decreased the number of dendrites/cell, but the decrease was not statistically significant (mStr+EGFP, 2.15 ± 0.10;
mStr-RACK1+EGFP, 2.15 ± 0.09; mStr+EGFP-Rab40C, 2.01 ± 0.07; and
mStr-RACK1+EGFP-Rab40C, 2.25 ± 0.07 [mean ± SEM]). Taken together, these results indicated that RACK1 has the ability to protect Varp protein from Rab40C and promotes forskolin-induced dendrite outgrowth of melanocytes.
DISCUSSION
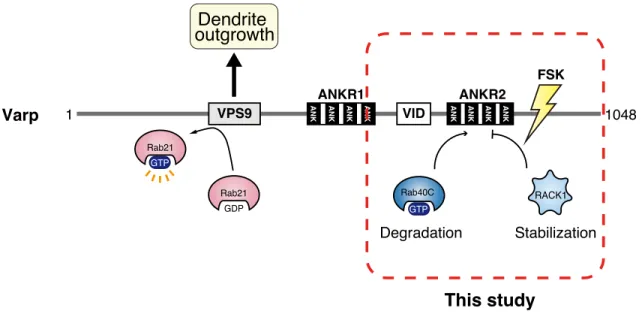
Varp has previously been reported to be regulated its protein expression level negatively by Rab40C that induces proteasomal degradation of Varp through interaction with the ANKR2 domain (Yatsu et al., 2015). However, the molecular mechanism by which Varp protein escapes from Rab40C remained unknown, and no attempt had ever been made to identify a positive regulator of the Varp protein expression level that counteracts the Varp degradation caused by Rab40C. In the present study, I identified RACK1 as a novel Varp ANKR2-domain-binding protein by yeast two-hybrid screening and demonstrated that forskolin, which increases the intracellular cAMP level by activating adenylate cyclase, increases the interaction between RACK1 and Varp (Figure 1). Because Varp has been shown to regulate forskolin-induced dendrite outgrowth of melanocytes (Ohbayashi et al., 2012b), I proceeded to investigate the functional involvement of RACK1 in this process by knockdown and overexpression experiments. The results showed that RACK1 is actually required for stabilization of Varp protein (i.e., for prevention of Varp proteasomal degradation) but does not affect the Varp mRNA level (Figure 2) and that knockdown of RACK1 inhibited forskolin-induced dendrite outgrowth (Figure 3), the same as Varp knockdown did (Ohbayashi et al., 2012b). More importantly, RACK1 has the ability to compete with Rab40C for binding to the Varp ANKR2 domain in vitro (Figure 4), and the inhibitory effect of Rab40C on dendrite outgrowth in forskolin-stimulated melanocytes was fully restored by simultaneous expression of RACK1 (Figure 5). These results taken together indicated that RACK1 is a positive regulator of the Varp protein expression level in melanocytes that directly competes with a negative regulator
Rab40C for binding to the ANKR2 domain (see model in Figure 7). The molecular mechanism by which the RACK1–Varp interaction is regulated by forskolin is completely unknown, but because RACK1 interacts with many kinases, including PKC, Src, and MAPK (Adams et al., 2011), phosphorylation is likely to regulate the interaction. Further research will be necessary to determine whether phosphorylation (or post-translational modification[s]) of RACK1 (or Varp) actually regulates the interaction between Varp and RACK1 or Rab40C during skin pigmentation.
Although in the present study I focused on the role of RACK1 in forskolin-induced dendrite outgrowth, RACK1 may also contribute to forskolin-independent functions of Varp, e.g., melanogenic enzyme transport, in melanocytes (Tamura et al., 2009; Tamura et al., 2011; Marubashi et al., 2016), because the RACK1–Varp interaction occurs even in the absence of forskolin (Figure 1d). Actually, RACK1 has previously been reported to be present on melanosomes in human primary melanocytes with an unknown mechanism and to regulate tyrosinase activity by recruiting PKCβ (Park et al., 2004). Moreover, knockdown of RACK1 in melan-a cells caused disappearance of tyrosinase signals from melanosomes without altering the tyrosinase mRNA level and decreased melanin content as well, the same as Varp knockdown did (Figure 8), suggesting that protection of Varp protein by RACK1 from Rab40C-mediated degradation is also crucial for melanogenic enzyme transport to melanosomes in melanocytes. Intriguingly, it has recently been reported that RACK1 heterozygous mice exhibit skin pigmentation defects characterized by a white belly spot and hypopigmented tail and paws, while RACK1 homozygous mice die at an early embryonic stage (Volta et al., 2013). The hypopigmented phenotype may be mainly attributable to defects in dendrite outgrowth (Figure 3)
and in melanin synthesis (Park et al., 2004; Figure 8), both of which are crucial for pigmentation and are regulated by Varp protein (Tamura et al., 2009; Ohbayashi et al., 2012b).
Because both RACK1 and Varp are expressed in non-melanocytic cells, RACK1-dependent stabilization of Varp protein may be retained in cells other than melanocytes and their interaction may be involved in retromer-dependent cargo transport pathways (Hesketh et al., 2014; McGough et al., 2014) and neurite outgrowth, both of which are positively regulated by Varp (Burgo et al., 2009; Burgo et al., 2012). It is noteworthy that RACK1 is highly expressed in mouse brain (Ashique et al., 2006) and has been shown to regulate neurite outgrowth through interaction with molecules other than Varp (Ensslen and Brady-Kalnay, 2004; Ceci et al., 2012; Dwane et al., 2014). Further investigation of the RACK1–Varp complex in neurite outgrowth will clarify the universal role of RACK1 in Varp protein stabilization.
In conclusion, the results of this study have revealed a novel function of the ANKR2 domain of Varp, i.e., RACK1-mediated stabilization of Varp protein in melanocytes. Based on my findings, I propose that the ANKR2 domain of Varp fine-tunes the level of Varp protein expression through interaction with either a positive regulator, RACK1 (this study), or a negative regulator, Rab40C (Yatsu et al., 2015) rather than directly regulating melanogenesis. Dysregulation of this process may lead to skin pigmentation defects, including impaired melanin synthesis and impaired dendrite outgrowth.
REFERENCES
Adams DR, Ron D, Kiely PA. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun Signal 2011;9:22.
Ashique AM, Kharazia V, Yaka R, Phamluong K, Peterson AS, Ron D. Localization of the scaffolding protein RACK1 in the developing and adult mouse brain. Brain Res 2006;1069:31–8.
Bennett DC, Cooper PJ, Hart IR. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer 1987;39:414– 8.
Beaumont KA, Hamilton NA, Moores MT, Brown DL, Ohbayashi N, Cairncross O, et al. The recycling endosome protein Rab17 regulates melanocytic filopodia formation and melanosome trafficking. Traffic 2011;12:627–43.
Burgo A, Sotirakis E, Simmler M-C, Verraes A, Chamot C, Simpson JC, et al. Role of Varp, a Rab21 exchange factor and TI-VAMP/VAMP7 partner, in neurite growth. EMBO Rep 2009;10:1117–24.
Burgo A, Proux-Gillardeaux V, Sotirakis E, Bun P, Casano A, Verraes A, et al. A molecular network for the transport of the TI-VAMP/VAMP7 vesicles from cell center to periphery. Dev Cell 2012;23:166–80.
Ceci M, Welshhans K, Ciotti MT, Brandi R, Parisi C, Paoletti F, et al. RACK1 is a ribosome scaffold protein for b-actin mRNA/ZBP1 complex. PLoS One 2012;7:e35034.
Cichorek M, Wachulska M, Stasiewicz A, Tymińska A. Skin melanocytes: Biology and development. Postepy Dermatol Alergol 2013;30:30–41.
advances. Traffic 2005;6:525–33.
Dwane S, Durack E, O’Connor R, O'Connor R, Kiely PA. RACK1 promotes neurite outgrowth by scaffolding AGAP2 to FAK. Cell Signal 2014;26:9–18.
Ensslen SE, Brady-Kalnay SM. PTPm signaling via PKCd is instructive for retinal ganglion cell
guidance. Mol Cell Neurosci 2004;25:558–71.
Fukuda M. Synaptotagmin-like protein (Slp) homology domain 1 of Slac2-a/melanophilin is a critical determinant of GTP-dependent specific binding to Rab27A. J Biol Chem 2002;277:40118–24.
Fukuda M, Kanno E. Analysis of the role of Rab27 effector Slp4-a/granuphilin-a in dense-core vesicle exocytosis. Methods Enzymol 2005;403:445–57.
Fukuda M, Kanno E, Mikoshiba K. Conserved N-terminal cysteine motif is essential for homo- and heterodimer formation of synaptotagmins III, V, VI, and X. J Biol Chem 1999;274:31421–7.
Gandin V, Senft D, Topisirovic I, Ronai ZA. RACK1 function in cell motility and protein synthesis. Genes Cancer 2013;4:369–77.
Hesketh GG, Pérez-Dorado I, Jackson LP, Wartosch L, Schäfer IB, Gray SR, et al. VARP is recruited on to endosomes by direct interaction with retromer, where together they function in export to the cell surface. Dev Cell 2014;29:591–606.
James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 1996;144:1425–36.
Kobayashi H, Etoh K, Marubashi S, Ohbayashi N, Fukuda M. Measurement of Rab35 activity with the GTP-Rab35 trapper RBD35. Methods Mol Biol. 2015;1298:207–216
for melanosome distribution in melanocytess. Mol Cell Biol 2003;23:5245–55. Li J-J, Xie D. RACK1, a versatile hub in cancer. Oncogene 2014;34:1890–8.
Loftus SK, Larson DM, Baxter LL, Antonellis A, Chen Y, Wu X, et al. Mutation of melanosome protein RAB38 in chocolate mice. Proc Natl Acad Sci USA 2002;99:4471–6.
Marubashi S, Shimada H, Fukuda M, Ohbayashi N. RUTBC1 functions as a GTPase-activating protein for Rab32/38 and regulates melanogenic enzyme trafficking in melanocytes. J Biol Chem 2016;291:1427–40.
McGough IJ, Steinberg F, Gallon M, Yatsu A, Ohbayashi N, Heesom KJ, et al. Identification of molecular heterogeneity in SNX27-retromer-mediated endosome-to-plasma-membrane recycling. J Cell Sci 2014;127:4940–53.
Ohbayashi N, Fukuda M. Role of Rab family GTPases and their effectors in melanosomal logistics. J Biochem 2012;151:343–51.
Ohbayashi N, Maruta Y, Ishida M, Fukuda M. Melanoregulin regulates retrograde melanosome transport through interaction with the RILP–p150Glued complex in melanocytes. J Cell Sci 2012a;125:1508–18.
Ohbayashi N, Yatsu A, Tamura K, Fukuda M. The Rab21-GEF activity of Varp, but not its Rab32/38 effector function, is required for dendrite formation in melanocytes. Mol Biol Cell 2012b;23:669–78.
Park HY, Wu H, Killoran CE, Gilchrest BA. The receptor for activated C-kinase-I (RACK-I) anchors activated PKC-b on melanosomes. J Cell Sci 2004;117:3659–68.
Raposo G, Marks MS. Melanosomes – dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol 2007;8:786–97.
receptor for protein kinase C: A homolog of the beta subunit of G proteins. Proc Natl Acad Sci USA 1994;91:839–43.
Schäfer IB, Hesketh GG, Bright NA, Gray SR, Pryor PR, Evans PR, et al. The binding of Varp to VAMP7 traps VAMP7 in a closed, fusogenically inactive conformation. Nat Struct Mol Biol 2012;19:1300–9.
Serrels B, Sandilands E, Frame MC. Signaling of the direction-sensing FAK/RACK1/PDE4D5 complex to the small GTPase Rap1. Small GTPases 2011;2:54–61.
Tamura K, Ohbayashi N, Maruta Y, Kanno E, Itoh T, Fukuda M. Varp is a novel Rab32/38-binding protein that regulates Tyrp1 trafficking in melanocytes. Mol Biol Cell 2009;20:2900–8.
Tamura K, Ohbayashi N, Ishibashi K, Fukuda M. Structure-function analysis of VPS9-ankyrin-repeat protein (Varp) in the trafficking of tyrosinase-related protein 1 in melanocytes. J Biol Chem 2011;286:7507–21.
Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet - Semin Med Genet 2004;131C:75–81.
Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: A model system to study vesicular trafficking. Pigment Cell Melanoma Res 2009;22:268–82.
Volta V, Beugnet A, Gallo S, Magri L, Brina D, Pesce E, et al. RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiency. Cell Mol Life Sci 2013;70:1439–50.
Wang F, Zhang H, Zhang X, Wang Y, Ren F, Zhang X, et al. Varp interacts with Rab38 and functions as its potential effector. Biochem Biophys Res Commun 2008;372:162–7.
Wei AH, Li W. Hermansky-Pudlak syndrome: Pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res 2013;26:176–92.
Wu X, Hammer JA. Melanosome transfer: It is best to give and receive. Curr Opin Cell Biol 2014;29:1–7.
Yatsu A, Shimada H, Ohbayashi N, Fukuda M. Rab40C is a novel Varp-binding protein that promotes proteasomal degradation of Varp in melanocytes. Biol Open 2015;44:267–75. Zhang X, He X, Fu X-Y, Chang Z. Varp is a Rab21 guanine nucleotide exchange factor and
regulates endosome dynamics. J Cell Sci 2006;119:1053–62.
Figure 1. RACK1 is a novel Varp-interacting protein.
(a) Schematic representation of Varp and its truncated mutants used in this study. (b) Schematic representation of RACK1 protein and its corresponding mRNA.
(c) Interaction of RACK1ΔN with Varp truncated mutants as revealed by yeast two-hybrid
ANKR2 ANKR1 N+VPS9 SC-LW SC-AHLW RACK1ΔN
a
b
c
1 317 105 1058 528 1099 bp 437 1095 bp Y2H #1 Y2H #3 714 1058 bp RACK1ΔN WD40 RACK1/Gnb2l1 ATG RACK1 mRNA stop WD40 WD40 WD40 WD40 WD40 WD40 1 1254 bpd
Varp 1 VPS9 ANK ANK ANK ANK VID ANK ANK ANK ANK 1048
ANKR1 ANKR2 N+VPS9 ANKR1 ANKR2 37 37 FLAG-EGFP FLAG-Varp 1 2 T7-RACK1 (kDa) IB : anti-T7 (Input) IB : anti-T7 (IP : anti-FLAG) IB : anti-FLAG (IP : anti-FLAG) 3 4 5 6
DMSOFsk TPA DMSOFsk TPA
150 100 75 50 37 25 2.0 3.0 1.0 0 DMSO Fsk TPA Bound RACK1 (a.u.) ∗
e
f
37 1 2 (kDa) IB : anti-Varp IB : anti-RACK1 3 anti-RACK1 IgG Input Control IgG150 100
assays.
(d) Interaction between T7-RACK1 and full-length FLAG-Varp. Before harvesting, the transfected COS-7 cells were stimulated for 3 hours with 20 µM forskolin (Fsk), 20 nM TPA, or DMSO alone. FLAG-EGFP was used as a negative control.
(e) Quantification of T7-RACK1 bands shown in the middle panel in d. The bars represent the means and SEM of three independent experiments. *, P<0.05 in comparison with DMSO (Dunnett’s test).
(f) Endogenous interaction between RACK1 and Varp (arrowhead) in FSK-treated mouse melanocytes (melan-a cells).
Figure 2. Knockdown of RACK1 in melanocytes decreases their level of Varp protein expression.
(a) Reduced expression of Varp in RACK1-KD melan-a cells as revealed by immunoblotting with the antibodies indicated.
(b) Quantification of Varp bands shown in the middle panel in a. The bars represent the means and SEM of six independent experiments. *, P<0.05; **, P<0.01 in comparison with the control (Dunnett’s test).
a
c
b
150 100 50 IB : anti-Varp IB : anti-β-actin Control siRNAst1 st2 RACK1 siRNA 1 2 3 (kDa) IB : anti-RACK1 37 25 1.2 1.0 0.8 0.6 0.4 0.2 0 Control RACK1 st1 RACK1 st2 Varp / β -actin (a.u.) ∗ siRNA: ∗∗d
1.4 1.0 0.8 0.6 0.4 0.2 0 Control RACK1 st1 RACK1 st2 RACK1 / GAPDH (a.u.) ∗∗ ∗∗ 2.5 2.0 1.5 1.0 0.5 0 Control RACK1 st1 RACK1 st2 Varp / GAPDH (a.u.) siRNA: siRNA: 1.2 100 37 IB : anti-Varp IB : anti-β-actin RACK1 siRNA st2 RACK1 siRNA st2 1 2 3 (kDa) IB : anti-RACK1 25 Control siRNA DMSO MG132(c) Knockdown efficiency of RACK1 siRNAs as revealed by real-time quantitative RT-PCR analysis. **, P<0.01 (Dunnett’s test).
(d) Exposure of RACK1-KD melan-a cells to MG132 or DMSO for 48 hours. Representative data from one of three independent experiments with similar results are shown.
Figure 3. Knockdown of RACK1 inhibits dendrite outgrowth of melanocytes.
(a) Melan-a cells were cotransfected with RACK1 siRNA (st1 or st2) or control siRNA and pEGFP-C1 (transfection marker) and then stimulated for 20 hours with 20 µM forskolin (Fsk) or DMSO alone. Images of EGFP-expressing cells were captured at random, and typical images of RACK1-KD cells (EGFP-expressing cells) are shown. Scale bars, 50 µm.
(b) Quantification of total dendrite length/cell shown in a. The bars represent the means and SEM of data from three independent experiments (totally 145 cells were analyzed). *, P<0.05; **, P<0.01 in comparison with the control (Dunnett’s test).
a
200 160 120 80 40 0 Control RACK1 st1 RACK1 st2 Control RACK1 st1 RACK1 st2 DMSO Fsk ∗Total dendrite length
/ cell ( μ m) ∗∗ ∗∗ ∗∗ DMSO Fsk Control st1 RACK1 siRNA
b
siRNA: st2Figure 4. RACK1 inhibits the interaction between Varp and Rab40C.
(a) COS-7 cells transiently expressing T7-Varp and FLAG-Rab40C (or FLAG-EGFP as a control) with or without HA-RACK1 were lysed, and the interaction between T7-Varp and FLAG-Rab40C was evaluated by immunoprecipitation with anti-FLAG tag antibody-conjugated agarose beads followed by immunoblotting with the antibodies indicated.
(b) Quantification of Varp bands shown in the second panel in a (normalized to the input Varp bands). The bars represent the means and SEM of data from four independent experiments. (c) The Varp protein level in RACK1-, Rab40C-, and RACK1/Rab40C-KD melan-a cells.
Marubashi et al., Fig. 4, Top
⇒
a
b
1.2 1.0 0.8 0.6 0.2 0 EGFP Bound Varp (a.u.) 0.4 - + + Rab40C ∗ HA-RACK1 : FLAG :c
37 FLAG-EGFP FLAG-Rab40C 1 2 T7-Varp (kDa) IB : anti-T7 (Input) IB : anti-T7 (IP : anti-FLAG) 37 3 HA-RACK1 IB : anti-FLAG (IP : anti-FLAG) IB : anti-HA (Input) + − + 150 100 150 100 150 100 50 IB : anti-Varp IB : anti-β-actinControlRACK1Rab40C
1 2 3 (kDa) IB : anti-RACK1 25 4 RACK1 + Rab40C siRNA:
d
1.4 1.2 0.8 0.6 0.4 0.2 0 Varp / β -actin (a.u.) siRNA: ∗ControlRACK1Rab40C RACK1
+ Rab40C
means and SEM of data from six independent experiments. *, P<0.05 (Student’s unpaired t-test).
EGFP mStr EGFP mStr EGFP mStr EGFP-Rab40C mStr EGFP mStr-RACK1 EGFP-Rab40C mStr-RACK1 DMSO Fsk
b
250 200 150 100 50 0 RACK1 MockTotal dendrite length
/ cell ( μ m) ∗∗ RACK1 Mock
RACK1 MockRACK1
Mock mStr:
EGFP: Mock Rab40C Mock Rab40C
a
Figure 5. RACK1 rescues Rab40C inhibition of dendrite outgrowth of melanocytes.
(a) Melan-a cells co-expressing EGFP-Rab40C (or EGFP alone) and mStr-RACK1 (or mStr alone) were stimulated for 20 hours with 20 µM forskolin (Fsk) or DMSO alone. Images of the cells expressing both EGFP-tagged and mStr-tagged proteins were captured at random, and typical images are shown. Scale bars, 50 µm.
(b) Quantification of total dendrite length/cell shown in a. The bars represent the means and SEM of data from one representative experiment (n=110 cells). **, P<0.01 (Tukey’s test).
Figure 6. RACK1 is unable to rescue Rab40C inhibition of dendrite outgrowth of Varp-KD melanocytes.
(a) Melan-a cells co-expressing EGFP-Rab40C and mStr-RACK1 (or mStr alone) or Varp-KD cells co-expressing EGFP-Rab40C and mStr-RACK1 were stimulated for 20 hours with 20 µM forskolin (Fsk). Images of the cells expressing both EGFP-tagged and mStr-tagged proteins were captured at random, and typical images are shown. Scale bars, 50 µm.
(b) Quantification of total dendrite length/cell shown in a. The bars represent the means and SEM of data from three independent experiments (totally 90 cells were analyzed). *, P<0.05; **, P<0.01 (Tukey’s test). RACK1 Control EGFP mStr EGFP-Rab40C mStr EGFP-Rab40C mStr-RACK1 Fsk
a
b
160 120 80 40 0Total dendrite length
/ cell ( μ m) ∗∗ RACK1 Mock mStr: EGFP-Rab40C ∗ EGFP-Rab40C mStr-RACK1 Varp siRNA Varp siRNA: - - +
Figure 7. A proposed model of the regulation of the Varp protein expression level by RACK1 and Rab40C in melanocytes.
In this model, Rab40C (negative regulator; Yatsu et al., 2015) and RACK1 (positive regulator; this study) competitively bind to the ANKR2 domain of Varp, and this competitive mechanism fine-tunes the level of Varp protein expression in melanocytes. Forskolin (Fsk) increases the interaction between RACK1 and Varp (Figure 1), and the stabilization of Varp protein contributes to promoting dendrite outgrowth of melanocytes.
Varp 1 VPS9 ANK ANK ANK ANK VID ANK ANK ANK ANK 1048
ANKR1 ANKR2 Rab40C GTP RACK1 Degradation Stabilization Rab21 GDP Rab21 GTP Dendrite outgrowth This study FSK
Tyrosinase Bright-field 120 100 80 60 40 20 0 Control RACK1 st1 RACK1 st2 Tyrosinase intensity (a.u.) ∗∗ ∗∗ Control st1 RACK1 siRNA st2 75 50 IB : anti-Tyrosinase IB : anti-β-actin Control siRNA st2st1 RACK1 siRNA 1 2 3 (kDa) IB : anti-RACK1 37 25
a
b
c
d
e
1.2 1.0 0.8 0.6 0.4 0.2 0 st2 Melanin / protein (a.u.) ∗∗ siRNA: siRNA: 1.2 1.0 0.8 0.6 0.4 0.2 0 Control RACK1 st1 RACK1 st2 Tyrosinase / GAPDH (a.u.) siRNA:Figure 8. Knockdown of RACK1 decreases the protein expression level of tyrosinase in melanocytes.
(a) Reduced expression of tyrosinase in RACK1-KD cells as revealed by immunoblotting with the antibodies indicated.
(b) Unaltered expression level of tyrosinase mRNA in RACK1-KD cells as revealed by real-time quantitative RT-PCR analysis.
(c) Reduced signals of tyrosinase in RACK1-KD melanocytes. Scale bars, 20 µm.
(d) Quantification of tyrosinase intensity shown in c. The bars represent the means and SEM of data from one representative experiment (n=120 cells). **, P<0.01 in comparison with the control (Dunnett’s test).
(e) Reduced melanin content in RACK1-KD melanocytes. The bars represent the means and SEM of data from five independent experiments. **, P<0.01 (Student’s unpaired t-test).
Chapter 2
Rab7B/42 is functionally involved in protein degradation on melanosomes
in keratinocytes
Abstract
Keratinocytes uptake melanosomes from melanocytes and retain them in the perinuclear region, where they form melanin caps. Although these processes are crucial to protecting nuclear DNA against ultraviolet injury, the molecular basis of melanosome uptake and decomposition in keratinocytes is poorly understood. One of the major reasons for its being poorly understood is the lack of a specific marker protein that can be used to visualize or monitor melanosomes (or melanosome-containing compartments) that have been incorporated into keratinocytes. In this thesis, I performed a comprehensive localization screening for mammalian Rab family small GTPases (Rab1~45) and succeeded in identifying 11 Rabs that were enriched around melanosomes that had been incorporated into keratinocytes. I also established a new assay by using a recently developed melanosome probe (called M-INK) as a means of quantitatively assessing the degradation of proteins on incorporated melanosomes in control and each of a series of Rab-knockdown keratinocytes. The results showed that knockdown or CRISPR/Cas9-mediated knockout of Rab7B (also identified as Rab42) in keratinocytes caused strong inhibition of protein degradation on melanosomes. My findings indicated that Rab7B/42 is recruited to melanosome-containing compartments and that it promotes protein degradation on melanosomes in keratinocytes without altering their melanin content.
Introduction
Melanosomes are specialized organelles that store melanin pigments in mammalian skin melanocytes (Raposo and Marks, 2007). Melanin is synthesized by melanogenic enzymes, e.g., tyrosinase and tyrosinase-related protein 1 (Tyrp1), and deposited within melanosomes. Mature darkly pigmented melanosomes are transported to the periphery of the melanocytes along the cytoskeleton, and they are ultimately transferred to surrounding keratinocytes (Boissy, 2003; Ohbayashi and Fukuda, 2012). After the melanosomes have been incorporated into the keratinocytes, melanin forms supranuclear caps (called “melanin caps”) that protect nuclear DNA against ultraviolet injury (Kobayashi et al., 1998). During the past few decades, a number of proteins involved in the biogenesis and transport of melanosomes in melanocytes have been identified (Ohbayashi and Fukuda, 2012; Bowman et al., 2019), and their molecular mechanisms and functions have gradually been elucidated (Serre et al., 2018). By contrast, the molecular mechanism of melanosome transfer from melanocytes to keratinocytes has remained elusive and is still a matter of controversy, although several transfer models and a related melanosome receptor have been proposed (Seiberg et al., 2000; Ando et al., 2012; Wu et al., 2012; Tarafder et al., 2014; and reviewed in Van Den Bossche et al., 2006; and Tadokoro and Takahashi, 2017). Moreover, subsequent processes such as accumulation and decomposition of the incorporated melanosomes in keratinocytes are poorly understood.
Actually, there have been conflicting reports on the fate or final destination of the melanosomes incorporated into keratinocytes. Some studies have reported that the melanosomes are degraded by autophagy (Murase et al., 2013; Yang et al., 2018; Kim et al.,
2019), while others have reported that they reside in nondegradative compartments (Correia et al., 2018; Hurbain et al., 2018). Although several proteins (e.g., LAMP1 [lysosomal-associated membrane protein 1], CD63, EEA1 [early endosomal antigen 1], Rab5B, and LC3) have been observed in melanosome-containing compartments (Murase et al., 2013; Correia et al., 2018; Hurbain et al., 2018), no specific marker protein for such compartments in keratinocytes has been identified, and no appropriate assay to assess the degradation of proteins on melanosomes in keratinocytes has been established.
In this chapter, I performed a comprehensive localization screening for mouse and human Rab family members (Matsui et al., 2011; Ishida et al., 2012) and succeeded in identifying 11 Rabs that were enriched around melanosomes that had been incorporated into keratinocytes. I then developed a new assay to quantitatively assess the degradation of proteins on such melanosomes in keratinocytes and found that depletion of Rab7B (also identified as Rab42; Itoh et al., 2006; referred to as Rab7B/42 below) in keratinocytes resulted in the strongest inhibition of protein degradation on incorporated melanosomes. These findings indicated that Rab7B/42 promotes protein degradation on melanosomes in keratinocytes.
Materials and Methods
MaterialsAnti-Rab7B/42 rabbit polyclonal antibody was produced by using purified GST-tagged mouse Rab7B/42 as described previously (Azouz et al., 2012; Homma et al., 2019). Anti-tyrosinase rabbit polyclonal antibody was prepared as described previously (Beaumont et al., 2011). The following antibodies used in this study were obtained commercially: anti-LAMP1 rat monoclonal antibody (clone: 1D4B) and anti-GM130 mouse monoclonal antibody (clone: 35) (BD Biosciences, San Jose, CA); anti-LBPA mouse monoclonal antibody (clone: 6C4; Echelon Biosciences, Salt Lake City, UT); anti-EEA1 rabbit monoclonal antibody (clone: C45B10; Cell Signaling Technology, Danvers, MA); anti-LC3 rabbit monoclonal antibody (PM036; MBL, Nagoya, Japan); anti-Nogo-A mouse monoclonal antibody (AHP1799; Bio-Rad); anti-Pmel mouse monoclonal antibody (clone: HMB45) (Dako North America, Carpinteria, CA); anti-Tyrp1 mouse monoclonal antibody (clone: Ta99) and anti-Myc mouse monoclonal antibody (clone: 9E10) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-Myc rabbit monoclonal antibody (C3956) and horseradish peroxidase-conjugated anti-FLAG tag (M2) mouse monoclonal antibody (Sigma-Aldrich, St. Louis, MO); anti-RFP rabbit monoclonal antibody (600-401-379; Rockland, Gilbertsville, PA), anti-β-actin mouse monoclonal antibody (G043; Applied Biological Materials, Richmond, BC, Canada); and Alexa Fluor-labeled secondary antibodies (Thermo Fisher Scientific, Waltham, MA). All other reagents used in this study were analytical grade or the highest grade commercially available.
pEGFP-C1 (Takara Bio Inc., Shiga, Japan) vectors carrying mouse Rab1A~43, pEF-FLAG-Rab7B/42, and pMRX-IRES-puro-EGFP-Rab7B/42 were prepared as described previously (Fukuda, 2003; Matsui et al., 2011; Etoh and Fukuda, 2019). The mouse Rab44 and Rab45 cDNAs were amplified from the Marathon-Ready adult brain and testis cDNAs (Takara Bio Inc.) by using specific pairs of oligonucleotides (sequence information available upon request) as described previously (Fukuda, 2003) and then subcloned into the pEGFP-C1 vector. The Rab nomenclature in this study is the nomenclature used in the National Center for Biotechnology Information (NCBI) database, and thus the names of several Rabs, e.g., Rab7B (previously Rab42), Rab42 (previously Rab43), and Rab43 (previously Rab41), differ from the names used in Itoh et al. (2006). The M-INK cDNA (Ishida et al., 2017) was subcloned into the pMRX-brs-Myc, pMRX-brs-EGFP, and pMRX-brs-mCherry vectors (Saitoh et al., 2003) (a kind gift from Shoji Yamaoka, Tokyo Medical and Dental University, Tokyo, Japan). Effective siRNAs against mouse Rabs used in this study except Rab44 siRNA (target sequence: 5’-GAGATCAGCTTGCTTTGA-3’) were prepared as described previously (Matsui and Fukuda, 2013).
Cell cultures and transfections
Black-mouse-derived melanocyte cell line melan-a (a generous gift from Dorothy C. Bennett, St. George’s Hospital Medical School, London, UK) (Bennett et al., 1987) and mouse keratinocyte cell line XB2 (purchased from ATCC) were cultured as described previously (Ishida et al., 2017). Plat-E cells (a kind gift from Toshio Kitamura, The University of Tokyo, Japan) and NIH3T3 cells were grown at 37°C in Dulbecco’s modified Eagle medium (044-29765; FUJIFILM Wako Pure Chemical, Osaka, Japan) supplemented with 10% fetal bovine serum, 100 U/ml penicillin G,
and 100 µg/ml streptomycin in a 5% CO2 incubator. An XB2 cell line stably expressing EGFP-Rab7B/42 or NIH3T3 cell lines stably expressing tagged (Myc, EGFP, or mCherry)-M-INK were established by retrovirus infection as described previously (Morita et al., 2000). Plasmids and siRNAs were transfected into XB2 cells by using Lipofectamine 2000 and Lipofectamine RNAiMAX (Thermo Fisher Scientific), respectively, each according to the manufacturer’s protocol.
CRISPR/Cas9-mediated Rab7B/42 KO in XB2 cells
The CRISPR/Cas9-mediated Rab7B/42 KO [single guide RNA target sequence: 5’-TCAGGAGCGGTTCCGCTCAA-3’] in XB2 cells was performed by using pSpCas9 (BB) 2A-Puro vector (Addgene, Cambridge, MA) and the procedure described previously (Mrozowska and Fukuda, 2016). Clonal lines were isolated and analyzed by immunoblotting with anti-Rab7B/42 antibody to verify the loss of endogenous Rab7B/42 protein expression. Rab7B/42 gene knockout was confirmed by sequencing genomic PCR products.
Preparation of melanosomes
Confluent melan-a cells (one 10-cm dish) were scraped off the culture dish, homogenized in a homogenization buffer (0.25 M sucrose, 10 mM HEPES-KOH, pH 7.2, 1 mM 2-mercaptoethanol, and 1 mM EDTA) by passing them through a 27-gauge needle 5 times, and then centrifuged at 500 × g for 1 minute. After collecting the supernatants and re-centrifuging at 10,000 × g for 3 minutes, the pellets were washed twice with the homogenization buffer and centrifuged again at 10,000 × g for 3 minutes. The pellets were then re-suspended in the homogenization buffer,
melanosome-rich solution was measured with a BioPhotometer D30 (Eppendorf, Hamburg, Germany) at 340 nm, and the melanosome concentration was calculated according to a calibration curve developed in-house (absorbance = 2.9876 × concentration [µg/µl] – 0.2854).
Preparation of M-INK-containing lysates
Confluent M-INK-expressing NIH3T3 cells (one 10-cm dish) were scraped off the culture dish, homogenized in 1 ml of PBS by passing through a 27-gauge needle 3 times, and centrifuged at 17,000 × g for 5 minutes. The supernatants were collected and used as M-INK-containing lysates (simply referred to as M-INK lysates below). The M-INK lysates were frozen with liquid nitrogen and stored at -80°C before use.
Uptake of melanosomes by keratinocytes
XB2 cells were cultured on 24-well plates containing a 15-mm diameter coverslip, and 50 µg of purified melanosomes were added per 500 µl of culture medium. After incubation for 48 hours, the cells were fixed with 4% paraformaldehyde. The coverslips were incubated with a 100 µl volume of M-INK lysates (containing complete EDTA-free protease inhibitor cocktail [Roche Applied Science, Penzberg, Germany] and 0.05% saponin), then with primary antibodies and lastly with appropriate Alexa Fluor-conjugated secondary antibodies. The samples were mounted using ProLong Diamond Antifade Mountant (Thermo Fisher Scientific) and examined for fluorescence with a confocal fluorescence microscope (FV1000D, Olympus, Tokyo, Japan).
M-INK degradation assay
µl), incubated for 10 minutes, and then centrifuged at 10,000 × g for 1 minute. The pellets were re-suspended with the culture medium and used immediately as M-INK-labeled melanosomes. After culturing XB2 cells with M-INK-labeled melanosomes, the cells were washed twice with PBS, collected by trypsinization, and centrifuged at 500 × g for 1 minute. The cell pellets were lysed in an SDS sample buffer (62.5 mM Tris-HCl pH 6.8, 2% 2-mercaptoethanol, 10% glycerol, and 0.02% Bromophenol Blue) and boiled. The lysates were subjected to SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Merck Millipore, Burlington, MA) by electroblotting. The blots were blocked with 5% skim milk in PBS containing 0.1% Tween-20 (FUJIFILM Wako Pure Chemical), incubated at room temperature with primary antibodies for 1 hour and then with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 hour. Immunoreactive bands were detected by using the ChemiDoc Touch imaging system (Bio-Rad, Hercules, CA) and quantified with ImageJ software (version 1.52b; National Institutes of Health).
Assay for degradation of melanosomal proteins in keratinocytes
XB2 cells were cultured on a 10-cm dish with or without 1 mg of purified melanosomes. After incubation for 36 hours, the cells were washed twice with PBS, collected by trypsinization, and centrifuged at 500 × g for 1 minute. The cell pellets were suspended in the homogenization buffer (containing complete EDTA-free protease inhibitor cocktail), homogenized by passing them through a 27-gauge needle 30 times, and then centrifuged at 3000 × g for 5 minutes. The pellets were lysed in the SDS sample buffer, vortexed with 0.5 mm glass beads for 30 minutes at 4°C, and boiled for 15 minutes. The lysates obtained were analyzed by immunoblotting with
Live-cell imaging
Live-cell fluorescence imaging was performed by using a FV1000D confocal fluorescence microscope with a 100 × oil/1.4 NA Plan Apochromatic objective lens and Fluoview software. XB2 cells stably expressing EGFP-Rab7B/42 were placed on a 35-mm glass bottom dish (MatTek, Ashland, MA) and incubated for 2 hours with mCherry-M-INK-labeled melanosomes before imaging. During live-cell imaging, the dish was mounted in a chamber (INUB-ONI-F2; Tokai Hit Co., Ltd., Shizuoka, Japan) to maintain incubation conditions at 37°C and 5% CO2.
Images were acquired at intervals of 10 minutes and analyzed with ImageJ software.
Melanin assay
Melanin content was assayed as described previously (Tamura et al., 2009). In brief, XB2 cells were cultured for 48 hours with melanosomes, washed two times with PBS, then cultured for 2 weeks in the absence of melanosomes. Their melanin content normalized to protein content was measured as optical density at 490 nm.
RESULTS
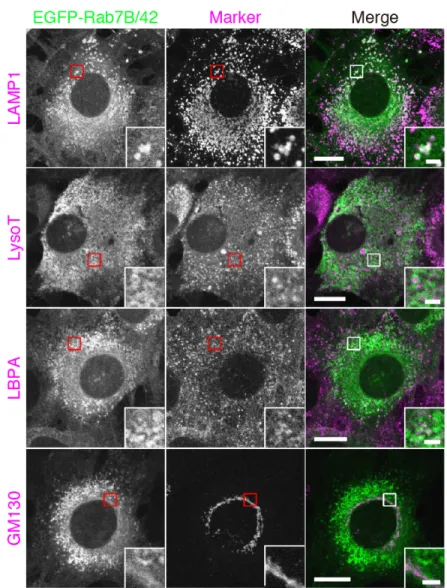
Melanosomes incorporated into keratinocytes are surrounded by LAMP1-positive and LysoTracker-negative structures
To identify a membrane compartment containing melanosomes in mouse XB2 keratinocytes, I co-stained XB2 cells that had been cultured with isolated melanosomes for 48 hours for various organelle markers and melanosomes. I stained for melanosomes by using the recently developed M-INK (Melanocore-INteracting Kif1c-tail) probe, which enables visualization of melanosomes in a fluorescent field (Ishida et al., 2017). Fluorescence visualization with M-INK is more accurate in the z-axis direction than bright-field observation, and M-INK makes it possible to correctly determine the intracellular position of melanosomes that have been incorporated into keratinocytes. Conventional markers for melanosomes (e.g., tyrosinase and Tyrp1) in melanocytes cannot be used, because they are not stained in keratinocytes that have incorporated melanosomes (Figure 1c; Ishida et al., 2017). The results of the co-staining analysis showed that many of the melanosomes were surrounded by LAMP1-positive structures (Figure 1a), but they were not well colocalized with other organelle markers, including EEA1 (early endosome marker), LBPA (lysobisphosphatidic acid; late endosome marker), LysoTracker Red (lysosome marker), LC3 (autophagosome marker) and Nogo-A (ER marker) (Figure 2). Because LAMP1 is known to localize both in lysosomes and late endosomes (Humphries et al., 2011), I also co-stained XB2 cells for LAMP1, LBPA (or LysoTracker Red), and melanosomes. Intriguingly, no M-INK-positive, LAMP1-positive structures were co-localized with LysoTracker Red or LBPA (Figure 1b, arrowheads). Such LAMP1-positive, LysoTracker-negative localization of melanosomes in keratinocytes has also recently been described in another study