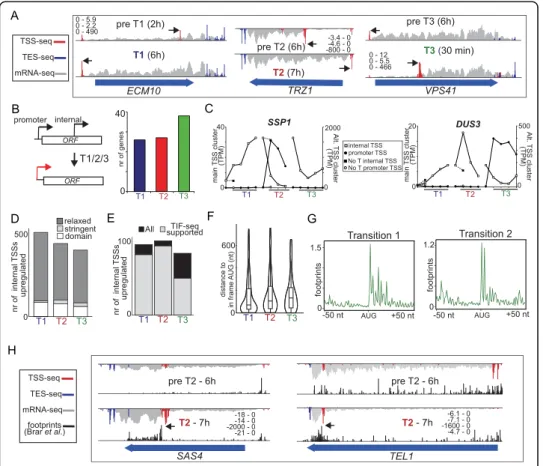
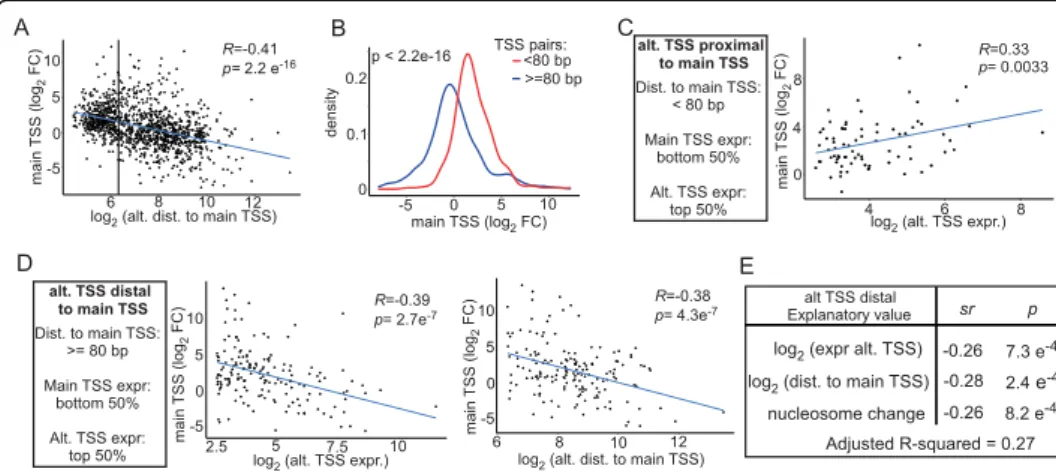
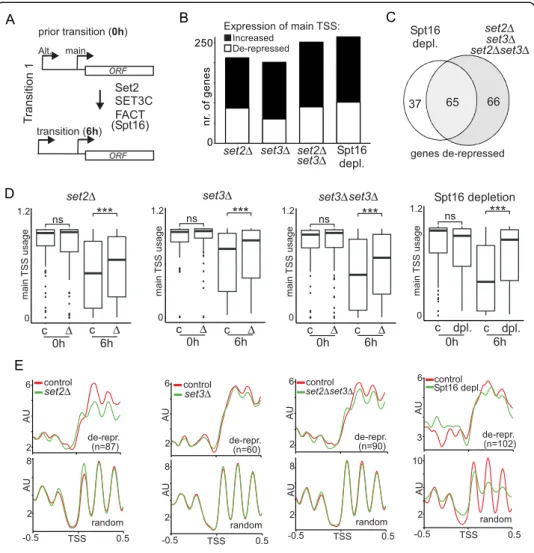
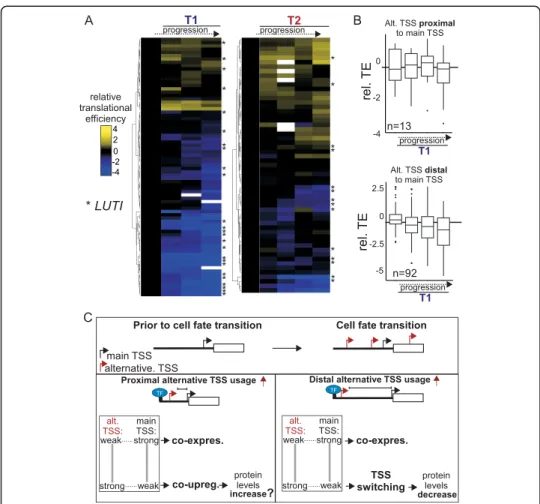
transcriptional tuning by alternative starts
Author Minghao Chia, Cai Li, Sueli Marques, Vicente Pelechano, Nicholas M. Luscombe, Folkert J.
van Werven journal or
publication title
Genome Biology
volume 22
number 1
page range 34
year 2021‑01‑14
Publisher BMC
Rights (C) 2021 The Author(s).
Author's flag publisher
URL http://id.nii.ac.jp/1394/00001759/
doi: info:doi/10.1186/s13059-020-02245-3
Creative Commons Attribution 4.0 International(https://creativecommons.org/licenses/by/4.0/)
R E S E A R C H Open Access
High-resolution analysis of cell-state
transitions in yeast suggests widespread transcriptional tuning by alternative starts
Minghao Chia 1,2 † , Cai Li 1,3 † , Sueli Marques 4 , Vicente Pelechano 4 , Nicholas M. Luscombe 1,5,6 and Folkert J. van Werven 1*
* Correspondence: folkert.
[email protected]
†
Minghao Chia and Cai Li contributed equally to this work.
1