九州大学学術情報リポジトリ
Kyushu University Institutional Repository
カイコの L セリン合成ならびに 1 炭素代謝に関す る分子基盤
モハメド, レズワヌル, ハクエ
https://doi.org/10.15017/2534505
出版情報:Kyushu University, 2019, 博士(農学), 課程博士 バージョン:
権利関係:
1
Studies on the molecular basis of de novo
L-serine biosynthesis and one-carbon metabolism in silkworm, Bombyx mori
Md. Rezwanul Haque
2019
2
Table of contents
Page
Chapter I. General introduction…... 4
Chapter II. Molecular characterization and expression analysis of the phosphoserine aminotransferase from Bombyx mori (bmPSAT)…….... 8
2.1. Introduction……….... 8
2.2. Materials and Methods……….. 10
2.3. Results………... 14
2.3.1. Cloning and sequencing of the bmPSAT cDNA……….. 14
2.3.2. Localization of the bmPSAT mRNA ……….……… 14
2.3.3.Functional characterization of bmPSAT……… 15
2.3.4.Amino acid composition in mulberry leaves………. 15
2.4. Discussion………. 17
Chapter III. Identification and characterization of the phosphoserine phosphatase from Bombyx mori (bmPSP)……….. 28
3.1. Introduction………... 28
3.2. Materials and Methods………. 30
3.3. Results………... 35
3.3.1. Cloning and sequencing of the bmPSP cDNA……….. 35
3.3.2. Localization of the bmPSP mRNA in various tissues……….. 35
3.3.3. Functional characterization of bmPSP……….. 36
3.3.4. Site-directed mutagenesis………. 36
3.4. Discussion……… 38
3
Chapter IV. Identification and characterization of the serine
hydroxymethyltransferase from Bombyx mori (bmSHMT)…….. …… 50
4.1. Introduction……… 50
4.2. Materials and Methods………... 52
4.3. Results……… 57
4.3.1. Cloning and sequencing of the bmSHMT cDNA encoding… 57 4.3.2. Expression of the bmSHMT mRNA ……….. 57
4.3.3. Overexpression and purification of bmSHMT and mutants……… 58
4.3.4.Biochemical and functional characterization of bmSHMT… 58 4.3.5. Amino acid residues involved in catalytic function…………. 59
4.4. Discussion………. 60
Chapter V. Identification and characterization of the 5,10 – methylenetetrahydrofolate dehydrogenase from the silkworm, Bombyx mori (bmMTHFD)………... 71
5.1. Introduction………... 71
5.2. Materials and Methods………. 73
5.3. Results………... 78
5.3.1. Cloning and sequencing of the bmMTHFD cDNA………….. 78
5.3.2. Distribution of the bmMTHFD mRNA ……… 78
5.3.3.Enzymatic properties of bmMTHFD……… 78
5.3.4. Amino acid residues involved in catalytic function…………. 79
5.4. Discussion………. 81
Chapter VI. General conclusion……….... 93
4
References……… 97
Abbreviations………. 113
Acknowledgements………. 115
5
CHAPTER I
General Introduction
Evolution of the capacity to produce essential metabolites, such as amino acids, is one of the most crucial biological processes because of its primal and important role in every living cell.
L-Serine is a nutritionally dispensable amino acid, acts as a basic structural unit of protein as well as a hub of one-carbon (1C) metabolism because of its role as a major 1C donor. Thus,
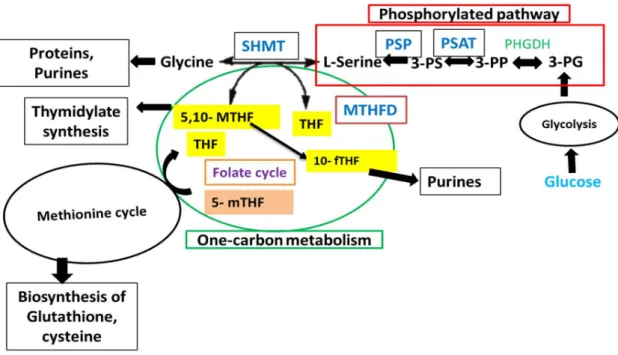
L-serine plays a crucial role in the synthesis of important cell constituent, including glycine, cysteine, thymidine, purines, and methionine in mammalian cells (Ducker & Rabinowitz, 2017). This neutral amino acid is also required for the production of sphingolipids, porphyrins, and neuromodulator D-serine (Snell, 1984; Snyder & Kim, 2000). Furthermore, Yoshida et al. (2004) reported that the de novo synthesis of L-serine is essential forembryonic or survival and tissue development in mammals. For de novo production of L-serine, two pathways are known to function. The first one is the phosphorylated pathway, wherein L- serine is produced from the glycolytic intermediate, D-3-phosphoglycerate (PGA) with the activity of three enzymes, D-3-phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase (PSAT), and phosphoserine phosphatase (PSP) (Fig. 1). This L-serine synthesis pathway was identified in bacteria, plants, and mammals (Snell, 1984; Reynolds et al., 1988; O’Gaora et al., 1989; van del Zel et al., 1989; Battchikova et al., 1996; Saito et al., 1997; Ho et al., 1998; Hester et al., 1999).
The other pathway operates through 1C metabolism, is a metabolic network that involves a set of enzymatic chemical reactions, where L-serine is synthesized in the reversal reaction
6
from glycine and 5,10-methylenetetrahydrofolate (MTHF) by the activity of 1C metabolic serine hydroxymethyltransferase (SHMT). This pathway is also required for the synthesis of amino acids (serine and glycine) and proteins, purine, and a vitamin by the action of SHMT along with another crucial enzyme methylenetetrahydrofolate dehydrogenase (MTHFD) (Linda & Jesse, 1991; Burda et al., 2015) (Fig. 1). This 1C metabolic network exists in the cytoplasm, mitochondria, and nucleus of eukaryotic cells (Linda & Jesse, 1991; Fox & Stover, 2008; Patrick, 2011).
Silkworm (Bombyx mori) is an economically important Lepidopteran insect that serves as a molecular genetic model for clarifying a wide range of biological issues. Silk fiber is the main economic part, which is synthesized in the silk gland of silkworm larvae.
Two proteins, fibroin and sericin, that are include high amounts of serine, glycine, and alanine, are the major constituents of silk fiber (Yamada, 1978; Takasu et al., 2002). Akai et al. (2005) reported that the posterior and middle parts of the silk gland of B. mori larvae secrete fibroin and sericin, respectively. The enzyme-mediated synthesis of L-serine is compatible with the prime characteristic of the production of sericin as well as other essential cell metabolites in B. mori. Keeping the above information in view, I tried to identify and characterize the enzymes that might be associated in the biosynthesis of L-serine, which acts as a central precursor of essential cell metabolites in silkworm. To the best of my knowledge, this is the first study to identify the mechanism of L-serine synthesis in silkworm as well as other insects. Therefore, the identification and biochemical characterization of enzyme activities are essential to reveal the mechanism of the production of L-serine, which, in turn, facilitates the biosynthesis of various cell metabolites in silkworm. The present study deals
7
with the identification, regional distribution, and characterization of the PSAT, PSP, SHMT, and MTHFD enzymes from B. mori.
8
Fig. 1 Schematic representation of the enzymatic reactions of one-carbon metabolism and phosphorylated pathway.
PG: phosphoglycerate; PP: phosphohydroxypyruvate; PS: phosphoserine; THF:
tetrahydrofolate; 5,10-MTHF: 5,10-methylene-THF; 10-fTHF: 10-formyl THF; 5-mTHF: 5- methyl-THF; PHGDH: phosphoglycerate dehydrogenase; PSAT: phosphoserine aminotransferase; PSP: phosphoserine phosphatase; SHMT: serine hydroxymethyltransferase; MTHFD: 5, 10-Methylenetetrahydrofolate dehydrogenase. Red box and green circle indicate the phosphorylated pathway and 1C metabolism, respectively.
9
CHAPTER II
Molecular characterization and expression analysis of a phosphoserine aminotransferase from Bombyx mori.
2.1. INTRODUCTION
PSAT (EC 2.6.1.52) is a PLP-dependent enzyme that catalyzes the reciprocal conversion of
L-glutamate and 3-phosphohydroxypyruvate (PHP) to 2-oxoglutarate and L-phosphoserine, which is crucial for L-serine synthesis in the phosphorylated pathway (Greenberg & Ichihara, 1957; Walsh & Sallach, 1966). In this pathway, three steps are involved in de novo L-serine production from the glycolytic intermediate, 3-phosphoglycerate (PGA). PSAT catalyzes the second step, whereas the first and the third steps are catalyzed by other two crucial enzymes, PHGDH (EC 1.1.1.95) and PSP (EC 3.1.3.3), respectively (Fig. 1), and this de novo L-serine production pathway is present in bacteria, plants, humans, and other animals (Snell, 1984;
Reynolds et al., 1988; O’Gaora et al., 1989; van del Zel et al., 1989, Battchikova et al., 1996;
Saito et al., 1997; Ho et al., 1998; Hester et al., 1999). In human PSAT (HsPSAT), the cDNA has two isoforms, named HsPSATα and HsPSATβ, generated by differential splicing (Baek et al., 2003). L-Serine is an standard amino acid that is not only required for protein formation, but also performs as a main 1C source that provides 1C units into the 1C metabolic pathway for the synthesis of important biomolecules, for example, glycine, cysteine, methionine, purines, and thymidine, sphingolipids, porphyrins, and neuromodulator D-serine in the living organisms (Snell, 1984; Snyder & Kim, 2000; Ducker & Rabinowitz, 2017).
10
The silk fiber, which is produced in the silk gland of B. mori, makes it an economically important insect. According to its function, a silk gland consists of three compartments, including the anterior (ASG), middle (MSG), and posterior silk glands (PSG).
The MSG and PSG are involved in the synthesis of the sericin and fibroin proteins of silk, respectively (Ishikawa & Suzuki, 1985; Akai et al., 1987; Sehnal and Akai, 1990). These two proteins include high content of serine, glycine, and alanine (Yamada, 1978; Takasu et al., 2002). Yamada (1978) reported that sericin comprises 29% serine, and fibroin comprises 44% glycine. Although serine is the most important amino acid in the silk gland, the de novo mechanism of L-serine synthesis in silkworm remains uncharted. Therefore, the identification and characterization PSAT from B. mori (bmPSAT) was carried out for the first time in this study.
11
2.2. MATERIALS AND METHODS
Insects
B. mori larvae (p50T strain) were reared on mulberry leaves at the Institute of Genetic Resources, Kyushu University (Fukuoka, Japan). The fat body was excised from the day-3 fifth-instar larvae in ice-cold 0.75% NaCl, rapidly frozen in liquid N2, and store at −80 °C prior to use.
Cloning and sequencing of the bmPSAT cDNA
The total RNA was prepared from the fat body of B. mori larvae by using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). For preparing the first-strand cDNA, the SuperScript II reverse transcriptase (Thermo Fisher Scientific, Carlsbad, CA) and an oligo-dT primer were used. The produced cDNA was used as a template for PCR amplification with the
following two oligonucleotide primers 5′-
CGCCTCGAGATGTCTAAAGTGTTTAATTTT-3′ (sense) and 5′-
CCGGATCCTTACTTGGAATGTTTTTTATAG-3′ (antisense). The primers were designed based on a sequence obtained from the SilkBase EST database (Mita et al., 2003). The underlined and double-underlined portion in primer sequence indicates the XhoI and BamHI restriction enzyme sites, severally. The Polymerase chain reaction (PCR) program included one cycle of initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 59 °C for 1 min, and extension at 72 °C for 2 min, followed by the final extension step at 72 °C for 10 min. After ligation with the pGEM-T Easy cloning vector (Promega, Madison, WI), the produced bmPSAT cDNA was transformed into Escherichia coli DH5α cells. The National Center for Biotechnology Information (NCBI;
12
https://www.ncbi.nlm.nih.gov) and Uni-Prot (https://www.uniprot.org) databases were used to retrieve the amino acid sequences of PSATs, and Clustal Omega were used for sequence alignment preparation. A phylogenetic tree was made by neighbor-joining (NJ) plot software (http://dua.prabi.fr/software/njplot).
Quantitative polymerase chain reaction (qPCR) analysis
The qPCR primers for bmPSAT and B. mori ribosomal protein 49 (Bmrp49) were sequenced.
bmPSAT F (forward): 5′-ATGGGATCCATGTCTAAAGTGTTTAATTTT-3′; bmPSAT R (reverse): 5′-CCAAGCTTTTACTTGGAATGTTTTTTATAG-3′; Bmrp49 F (forward): 5′- GATGTGTTTTATATTC-3′: Bmrp49 R (reverse): 5′-GCATCATCAAGATTTCCAGCTC- 3′ were used as primer sequences. The qPCR was performed in a 20 μl final volume consisted of 10 μl SYBR® Green reagents, 1 μl (100 ng/μl) cDNA, 1.6 μl (800 nM) each of forward and reverse primers, and 5.8 μl distilled water. The qPCR amplification began with an initial denaturation step at 95 °C for 1 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min, and extension at 60 °C for 1 min. The expression level was standardized against the similar Bmrp49 levels and calculated as the bmPSAT/Bmrp49 ratio.
The data from three experimental replicates were used for calculation.
Overexpression and purification of bmPSAT
The bmPSAT clone was digested with XhoI and BamHI, ligated into the expression vector pCold‐SUMO‐M (HaiGene, Chenghaerbin, China) and transformed into competent E. coli Rosetta (DE3) pLysS cells (Merck Millipore, Danver, MA). The cells were then cultured at 37 °C in 250 mL of LB medium containing 250 µl ampicillin (50 mg/ml) until OD600 become
13
approximately 0.50. Subsequently, the culture was cooled on ice for 30 min. After the addition of 300 µl of 1 mM isopropyl thio-β-d-galactoside (IPTG) as inducer, it was cultured overnight at 20 °C. The bacterial cells were harvested by centrifugation at 3,500 rpm and saved at −20 °C prior to use. The cell pellet was resuspended in 1X phosphate-buffered saline (PBS) buffer (pH 7.0) containing 0.1 mg/mL lysozyme. After incubating at 37 °C, the cells were disrupted by sonication. The supernatant was clarified by centrifugation at 14,000 rpm and subjected to Ni2+-affinity chromatography (His-Pur Ni-NTA resin, Thermo Scientific, Meridian Rd., Rockford, U.S.A). After washing with 1X PBS, 1X PBS with 0.5 M imidazole was used for eluting the protein. The resulting recombinant protein was further purified by gel filtration and its purity was examined by performing sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE) using 15% polyacrylamide gel with 0.1%
SDS (Laemmli, 1970). The protein gel was stained with Coomassie brilliant blue (CBB) R250.
Enzymatic assay of bmPSAT
The enzymatic activity of bmPSAT was assayed spectrophotometrically. The enzyme activity for forward reaction was measured by coupling the phosphoserine aminotransferase with glutamate dehydrogenase at 25 °C. A decrease in absorption was observed at 340 nm in a Nano Drop® ND-1000 spectrophotometer. For determining the optimum pH, the Britton-Robinson buffer (pH 2.0−12.0) was used. The activity assay was accomplished by using 10 μg of enzyme in 50 μl of reaction mixture consisting 8 mM glutamic acid, 32 mM ammonium acetate, Britton-Robinson buffer (pH 8.0), 0.18 mM NADPH, 20 μM PLP, and 12 U glutamate dehydrogenase. The reaction was initiated by adding of 0.5 mM PHP.The
14
kinetic parameters were assessed using various amounts of glutamate (4–12 mM) and PHP (0.1–0.6 mM). The Km and Vmax were estimated with the Line-weaver Burk plots. The initial velocities were computed using the molar extinction coefficient of NADPH (6.22 mM-1 cm-
1).
Analysis of amino acids in mulberry leaves
Mulberry leaves were taken from the plants grown at the farm of the Institute of Genetic Resources, Kyushu University Graduate School (Fukuoka, Japan). The leaves were rapidly frozen in liquid N2 and stored at −80°C prior to use. The homogenized leaves were centrifuged at 15,000 rpm for 30 min at 4°C. he mixture of supernatant and 1/10 volume of 5% perchloric acid (HClO4) was incubated on ice for 25 min, and then centrifuged for 20 min at 15,000 rpm at 4°C. 1/10 volume of 8 N KOH was used for neutralization of supernatant. The amino acid composition of the supernatant was determined by an Acquity UPLC H-class system (Waters, Milford, MA).
15 2.3. RESULTS
2.3.1. Cloning and sequencing of the bmPSAT cDNA
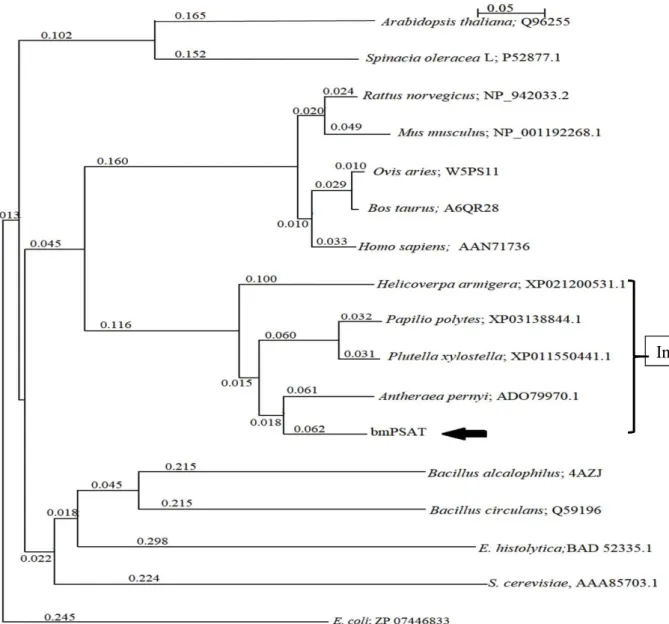
The obtained bmPSAT cDNA encoding 364 amino acids (Fig. 2.1) with a computed Mw and the isoelectric point (pI) 40,199 and 6.97, respectively. The sequence alignment exposed that bmPSAT shared 58%, 87%, 47%, 50%, and 48% identity with the PSAT of Homo sapiens (HsPSAT), Antheraea pernyi, Bacillus circulans, Arabidopsis thaliana, and E. coli, respectively (Fig.2.1). This result showed that bmPSAT was more closely related to the PSAT enzyme from insects, humans, and bacteria because of the presence of almost similar amino acid inserts and lack of N-terminal sequence. The bmPSAT protein consists of the postulated signature sequence for the enzyme PSAT. In bmPSAT, all crucial amino acids present in the functional sites within the substrate- and cofactor-binding domains, were conserved, as predicted from the crystal structure of the E. coli PSAT (Fig. 2.1). The phylogenetic tree analysis showed that bmPSAT was evolutionarily close to the PSAT of insects (Fig.2.2).
2.3.2. Localization of the bmPSAT mRNA
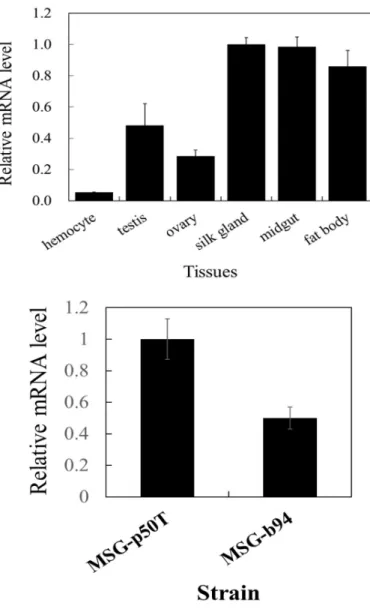
The distribution of the bmPSAT mRNA obtained from various tissues was determined by qPCR. The result showed all tested tissues of silkworm contained the bmPSAT mRNA, even though the relative levels were widely ranging. The bmPSAT mRNA was high in the silk gland and midgut, lower in the fat body (14% less than that in the silk gland), ovary (72%
less than that in the silk gland), testis (77% less than that in the silk gland), and the lowest in the hemocyte (95% less than of silk gland) (Fig. 2.3.A). The analysis of the bmPSAT mRNA
16
between the MSGs of the standard silkworm (p50T strain) and sericin-deficient b94 strain showed a reduction of approximately 50% expression in the b94 strain (Fig. 2.3.B)
2.3.3. Functional characterization of bmPSAT
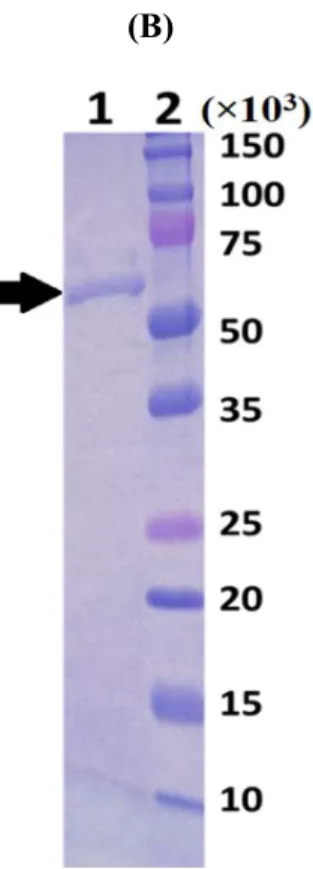
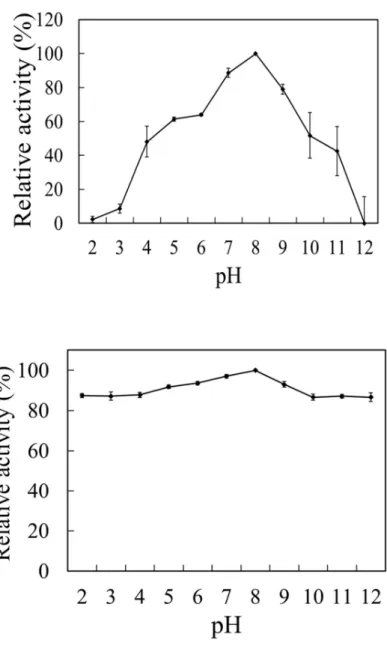
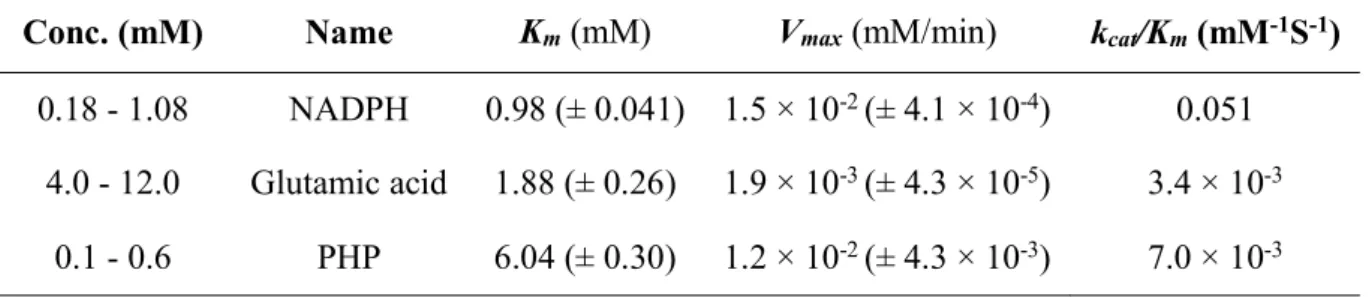
bmPSAT was successfully overproduced in E. coli (Fig. 2.4.A). The produced fused protein contained bmPSAT, a small ubiquitin-related modifier (SUMO), and a 6X His-tag. The optimal results for the purification of bmSHMT by a nickel column and gel filtration would show only one band of protein at 56,000 in the elution fraction. (Fig. 2.4.B), that included approximately 16,000 of SUMO and His-tag. The enzyme utilizes glutamic acid and PHP preferentially for transamination. The supreme activity of bmPSAT was found at pH 8.0 (Fig.
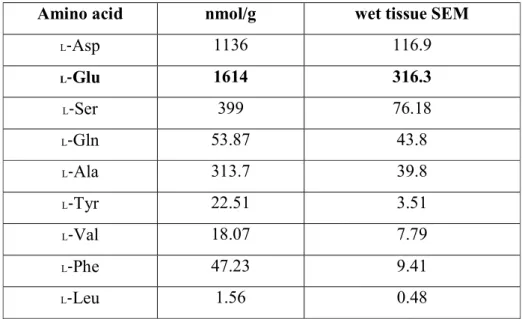
2.5.A), and the enzyme showed activity at almost all pH levels; more than 80% of the highest activity was achieved at pH 2.0–12.0 (Fig. 2.5.B), and the activity was not stable at temperatures >25 °C (data not shown). The activity of the enzyme was appraised at optimal temperature and pH, indicating that the Km, Vmax,and kcat/Km values were 0.98 (± 0.041) mM, 1.5 × 10-2 (± 4.1 × 10-4) mM min-1, and 0.051 mM-1S-1 for NADPH and was 1.88 (± 0.26) mM, 1.9 × 10-3 (± 4.3 × 10-5) mM min-1, and 3.4 × 10-3 mM-1S-1 for glutamic acid, respectively. Moreover, the Km, Vmax,and kcat/Km values toward PHP were 6.04 (± 0.30) mM, 1.2 × 10-2 (± 4.3 × 10-3) mM min-1, and 7.0 × 10-3 mM-1S-1, respectively (Table 2.1).
2.3.4. Amino acid composition in mulberry leaves
The amino acid composition of mulberry leaves is presented in Table 2.2. It was found that mulberry leaves contained the highest amount of L-Glu compared to other amino acids. The
17
amounts of amino acids in mulberry leaves followed the order L-Glu > L-Asp >L-Ser > L-Ala
> L-Gln > L-Phe > L-Tyr > L-Val > L-Leu.
18
2.4. DISCUSSION
I have identified and shown functional properties of the B. mori PSAT, which is crucial for the formation of L-serine in the phosphoserine synthetic pathway. The bmPSAT cDNA consisted of364 amino acids. The theoretical molecular mass of this enzyme was 40,000, which was similar to the PSAT isolated from Scenedesmus obliquus (40,000), Entamoeba histolytica and mammals (3,6000) (40,000) (Stolz et al., 1994; Baek et al., 2003; Vahab &
Nozaki, 2006), but lower than that isolated from the sheep brain (96,000) (Hirsch &
Greenberg, 1967). The deduced amino acid sequences were homologous to the PSATs from Homo sapiens (HsPSAT), A. pernyi, B. circulans, A. thaliana, and E. coli. The highest identity (87%) was observed incase of PSAT isolated from A. pernyi (Chinese oak silk moth).
A BLAST search (http://metazoa.ensembl.org/Bombyx_mori/Info/Index) showed that bmPSAT exists as a single gene in silkworm; however, this enzyme has two forms in humans (PSATα and PSATβ)(Baek et al., 2003). The lysine residue acts as the PLP-binding site, and the residues around the PLP-binding site have been determined previously (Ouzounis et al., 1993; Belhumeur et al., 1994; Bairoch et al., 1996; Saito et al., 1997). In addition, the multiple sequence alignment revealed that all important residues associated with the active site of bmPSAT were conserved, indicating that bmPSAT functions through substrate and cofactor binding. The proposed signature sequence for PSAT (Belhumeur et al., 1994; Saito et al., 1997; Baek et al., 2003; Wang et al., 2013) was also found in the bmPSAT protein. The qPCR analysis of the RNA isolated from the silkworm tissues revealed that the bmPSAT mRNA is widely distributed. The higher amount mRNA was observed in the silk gland, midgut, and fat body of silkworm. Silk gland is the main organ that produces
19
silk fiber, which mainly comprises sericin and fibroin. This result indicates that bmPSAT plays a role in the production of L-serine, which is the main constituent of sericin. PSAT is responsible for the biosynthesis of L-serine, which plays a role in the formation of important cell metabolites, including glycine, cysteine, methionine, purines, thymidine, neuromodulator D-serine, and sphingolipids in mammals (Snell, 1984; Snyder & Kim, 2000).
Thus, the result of mRNA localization showed that bmPSAT could also play a role in the production of those cell metabolites in the larvae of silkworm. It was found that L-Glu has the highest concentration among others in mulberry leaves. Higher amount of the bmPSAT mRNA in the midgut revealed that bmPSAT uses L-Glu as a substrate, which is supplied through mulberry leaves upon being fed by silkworm, for the biosynthesis of L-serine.
In this study, I compared the levels of the bmPSAT mRNA between the MSGs of the wild-type silkworm (p50T) and sericin-deficient variant b94. Interestingly, it was observed that the level of bmPSAT mRNA is low (50% of the standard p50T) in the b94 strain compared to the p50T strain (Fig. 2.3.B). This result provided evidence of the reduced expression of bmPSAT mRNA in the MSG of b94, one of the crucial factors for sericin deficiency in that strain. The bacterially produced soluble bmPSAT showed the highest activity at pH 8.0, which was similar to the activities of the PSATs obtained from humans, sheep brain, and Entamoeba histolytica (Hirsch & Greenberg, 1967; Baek et al., 2003; Vahab
& Nozaki, 2006), but was different from the activities of the PSATs obtained from Bacillus alcalophilus and Bacillus circulans (Dubnovitsky et al., 2005; Kapetaniou et al., 2006). The determination of the kinetic constants revealed that the glutamate Km value (1.88 mM) of bmPSAT was higher than that of the PSATs isolated from Bacillus circulans (1 mM) and E.
histolytica (0.778 mM) (Kapetaniou et al., 2006; Vahab & Nozaki, 2006), but lower than that
20
of the PSAT isolated from A. thaliana (5.05 mM) (Vahab & Nozaki, 2006). However, this result was consistent with the PSAT isolated from the bovine serum (1.2 mM) (Basurko et al., 1999). Furthermore, I found that the Km value for PHP higher in B. mori than in other organisms, suggesting that bmPSAT has low affinity to this substrate. Therefore, the kinetic results showed the capacity of bmPSAT to recognize glutamate and PHP as substrates and NADPH as a cofactor. Besides, this was in agreement with the conservation of the substrate- binding and PLP-binding sites in bmPSAT (Fig. 2.1).
21
Fig. 2.1. Multiple alignment of deduced amino acid sequence of PSAT.
The sequences of PSATs from various organisms were collected from the BLAST search and Uni-Prot database: bmPSAT (determined in the study), Homo sapiens (HsPSAT;
AAN71736.1), A. pernyi (ADO79970.1), E. coli (ZP_07446833.1), B. circulans (Q59196), andA. thaliana (Q96255). Red and green boxes indicate the glutamate and cofactor PLP- binding sites, respectively. PLP: Pyridoxal 5'-phosphate; PSAT: Phosphoserine aminotransferase. The signature sequences for PSAT proteins are underlined.
22
Fig. 2.2. Phylogenetic tree of PSATs.
The phylogenetic tree was prepared with the NJ plot software using various PSATs sequence gathered from the NCBI and Uni-Prot databases. The species names and accession numbers are given in the figure. The arrow represents the phylogenetic position of bmPSAT. E. coli (ZP_07446833) was used as outgroup. Scale bar (0.05) shows number of amino acid substitutions per site.
Insects
23
Fig. 2.3.
Localization of bmPSAT mRNA.(A) Localization of the bmPSAT mRNA in different tissues of standard silkworm (p50T). (B) Analysis of the bmPSAT mRNA between the MSGs of the p50T and b94 strains. We performed a qPCR to detect the bmPSAT mRNA expression levels in different tissues. The amounts of mRNA were analyzed by qPCR as described in the Materials and Methods. The error bars represent the standard error obtained from three experiments.
(B) (A)
24
Fig. 2.4. (A) Overexpression and (B) Purification
(A) Overexpressed bmPSAT. Lane 1 represents the protein molecular size marker;
lane 2 shows the band corresponding to bmPSAT without induction with 1 mM IPTG, whereas lane 3 shows the band corresponding to the bmPSAT protein overexpressed using 1 mM IPTG as an inducer. The arrow indicates the overexpressed bmPSAT band, and (B) SDS-PAGE of the bmPSAT protein purified using Ni2+
chromatography. The bmPSAT protein was purified as described in Materials and Methods. Lane 1 is the fraction that eluted from Ni2+-resin, and Lane 2 shows protein molecular size marker.
(A) (B)
25
Fig. 2. 5. (A) Optimum pH and (B) pH stability
(A) Enzyme activity was assayed by using Britton-Robinson buffer with pH 8.0 at 25°C and (B) bmPSAT was pre-incubated at pH 2.0–12.0 at 4 °C for 24 h, and its net activity was assayed at temperature 25°C and pH 8.0. The maximal value was set to 100%.
(A)
(B)
26
Table 2.1. (a)
Kinetic properties of bmPSAT towards NADPH, glutamic acid, and PHP.Line-weaver Burk plot was used for calculating the kinetic parameters
.
Conc. (mM) Name Km (mM) Vmax (mM/min) kcat/Km (mM-1S-1) 0.18 - 1.08 NADPH 0.98 (± 0.041) 1.5 × 10-2 (± 4.1 × 10-4) 0.051
4.0 - 12.0 Glutamic acid 1.88 (± 0.26) 1.9 × 10-3 (± 4.3 × 10-5) 3.4 × 10-3 0.1 - 0.6 PHP 6.04 (± 0.30) 1.2 × 10-2 (± 4.3 × 10-3) 7.0 × 10-3
27
Table 2.1. (b)
Amino acid composition of mulberry leaves. The amino acid composition of mulberry leaves was analyzed by UPLCAmino acid nmol/g wet tissue SEM
L-Asp 1136 116.9
L-Glu 1614 316.3
L-Ser 399 76.18
L-Gln 53.87 43.8
L-Ala 313.7 39.8
L-Tyr 22.51 3.51
L-Val 18.07 7.79
L-Phe 47.23 9.41
L-Leu 1.56 0.48
28 Acknowledgements
I would like to thank Ayumi Koyanagi, Takashi Ichinose, Maiko Abiru, Shinya Mohri (Students of Professor Dr. Shigeki Furuya laboratory), who were done the experiment on amino acids composition in mulberry leaves and analysis of bmPSAT mRNA expression between middle silk gland of standard silkworm (p50T) and b94 strain in this chapter. Also, I am thankful for Dr. Kohji Yamamto to excise fat body, clone, and sequence bmPSAT, and for Aiko Hirowatari to prepare expression vector for bmPSAT.
29
CHAPTER III
Identification and characterization of the phosphoserine phosphatase from B. mori
3.1. INTRODUCTION
Phosphoserine phosphatase (PSP;EC: 3.1.3.3) is an Mg2+-dependent enzyme that catalyzes the final step of L-serine anabolism by dephosphorylation of phosphoserine in the phosphorylated pathway (Chiba et al., 2012). In this pathway, PSP acts as a key enzyme in catalyzing the final third step of L-serine biosynthesis, wherein the first and the second step are catalyzed by PHGDH and PSAT respectively. In the previous chapter (chapter II), I have identified and characterized the second step catalyzing bmPSAT, which is involved in the biosynthesis of phosphoserine, which then acts as a substrate of PSP. Bridgers (1965) reported that beside other pathways, the phosphorylated pathway is the major pathway that produces more than 90% of L-serine in living cells. Metalloproteins, including nucleases, kinases, and phosphatases, use metal ions as essential active site components for their catalytic activity. So far, one the basis of metal requirement, two categories of PSPs have been studied. The first one is Mg2+-dependent PSP (dPSP) that belongs to the haloacid dehalogenase-like hydrolase (HAD) superfamily (Neuhaus & Byrne, 1958; Borkenhagen &
Kennedy, 1959; Koonin & Tatusov, 1994) and the other one is metal-independent PSP (iPSP), which exists in autotrophic bacteria (Chiba et al., 2012; Kim et al., 2016; Kim et al., 2017).
This enzyme is widely distributed from bacteria to humans. In the present study, the Mg2+-
30
dependent PSP, which catalyzesan irreversible reaction leading to the production of L-serine and inorganic phosphate, was identified. L-Serine is responsible for the biosynthesis of important cell constituents, including glycine, cysteine, methionine, purines, proteins, thymidine, sphingolipids, porphyrins, and neuromodulator D-serine in breathing cells, besides acting as a hub of 1C metabolism because of its role as a main 1C donor (Snell, 1984;
Ducker & Rabinowitz, 2017; Snyder & Kim, 2000). Silkworm produces silk fiber in the silk gland. Two type of silk proteins, fibroin and sericin, have been identified as the major constituents of silk cocoons. Yamada (1978) reported sericin contains a high content of serine (approximately 30%), and fibroin contains a high content of glycine (approximately 44%).
The PSP-mediated L-serine synthesis is in agreement with the fact that serine has the highest content in silk. The identification and characterization of SHMT and5,10-MTHFD, which are involved in the 1C metabolism to produce essential cell metabolites in silkworm, have been described in Chapter IV and V. The L-serine produced via phosphorylated pathway is converted to glycine by SHMT in the silk gland as well as other organs of silkworm. Several studies on PSP with reference to its metabolic significance have already been demonstrated, but to our knowledge, no study on this enzyme has been reported in silkworm. Therefore, the identification and biochemical conformation of PSP activity are essential to reveal the production of L-serine that facilitates the biosynthesis of various cell metabolites in silkworm.
The present chapter deals with the identification, regional distribution, and characterization of PSP in B. mori.
31
3.2. MATERIALS AND METHODS
Insects
B. mori larvae (p50T strain) were reared on mulberry leaves at the Institute of Genetic Resources, Kyushu University (Fukuoka, Japan). The fat body was excised from the day-3 fifth-instar larvae in ice-cold 0.75% NaCl, rapidly frozen in liquid N2, and stored at −80 °C prior to use.
Cloning and sequencing of the bmPSP cDNA
Total RNA was isolated from the fat body of B. mori larvae using the Sepasol-RNA 1 (Nacalai Tesque), according to the manufacturer’s protocol. The first strand of cDNA was obtained using the SuperScript II First-Strand Synthesis System for RT-PCR F (Thermo Fisher Scientific, Carlsbad, CA) and an oligo-dT primer. Thereafter, the cDNAs were amplified by PCR using the following two oligonucleotide primers 5′-
GCGCATATGGCTCTTTACAGTTTGAAAAGC-3′ (sense) and 5′-
ATTGGATCCTTACCAGGCTGCCTTCTTCTT-3′ (antisense). These primers were designed based on a sequence found from the SilkBase EST database (Mita et al., 2003). The Ndel and BamHI restriction enzyme sites are depicted by the underlined and double- underlined areas in the primer sequences. The PCR amplification was performed using the following reaction program: one cycle of initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 59 °C for 1 min, and extension at 72 °C for 2 min, and a final extension step at 72 °C for 10 min. The produced bmPSP cDNA was directly inserted into the pGEM-T Easy cloning vector (Promega, Madison, WI), which
32
was then transformed to E. coli DH5α cells. The NCBI and Uni-Prot databases were used to obtain the amino acid sequences of PSPs from various organisms, and a multiple sequence alignment was prepared through Clustal Omega. The NJ plot software (http://dua.prabi.fr/software/njplot) was used to construct a phylogenetic tree.
qPCR analysis
A qPCR was performed to confirm the presence of the mRNA expression profiles in bmPSP.
The SuperScript II First-Strand Synthesis System was used for synthesizing cDNAs from mRNAs. The housekeeping gene Bmrp49 was used as an internal control for cDNA normalization. The primers for bmPSP and B. mori ribosomal protein 49 (Bmrp49) were designed. bmPSP F (forward): 5′-CAGGCCTAACGTCGGTCAGA-3′; bmPSP R (reverse):
5′-GACTCCTGAATCCGCCCGAA-3′; Bmrp49 F (forward): 5′-GATGTGTTTTATATTC- 3′; Bmrp49 R (reverse): 5′-GCATCATCAAGATTTCCAGCTC-3′ were used as primer sequences. The qPCR was performed in a total volume of 20 μl containing 10 μl SYBR®
Green reagents, 1 μl (100 ng/μl) cDNA, 1.6 μl (800 nM) each of forward and reverse primers, and 5.8 μl distilled water. The qPCR amplification began with an initial denaturation step at 95 °C for 1 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min, and extension at 60 °C for 1 min. The expression level was standardized against the Bmrp49 expression level and set as the bmPSP/Bmrp49 ratio. The samples were examined in triplicate.
Overexpression and purification of bmPSP
33
The bmPSP clone was digested with NdeI and BamHI, ligated into the expression vector pET-15b (Merck Millipore, Darmstadt, Germany), and transformed into competent E. coli Rosetta (DE3) pLysS cells (Merck Millipore). The transformant was at 37 °C in 250 mL of LB medium containing 250 µl ampicillin (50 mg/ml) until OD600 become approximately 0.50–0.60. Thereafter, the culture was cooled on ice for 30 min and then incubated overnight at 20 °C for the induction of protein expression by adding 300 µl of 10 mM IPTG.
Subsequently, the cells were harvested by centrifugation at 3,500 rpm and stored at −20 °C until use. The cell pellet was resuspended in 20 mM HEPES; pH 7.0 buffer containing 0.3 M NaCl, 10 mM imidazole (pH 7.0), and 0.1 mg/mL lysozyme. After incubating at 37 °C, the cells were disrupted by sonication. The supernatant was clarified by centrifugation at 14,000 rpm and subjected to Ni2+-affinity chromatography (His-Pur Ni-NTA resin, Thermo Scientific, Meridian Rd., Rockford, U.S.A). After washing the chromatography column with 20 mM HEPES containing 0.3 M NaCl and 25 mM imidazole buffer, 20 mM HEPES; pH 7.0 containing 0.3 M NaCl with 0.5 M imidazole was used for eluting the protein. The resulting recombinant protein was further purified by gel filtration, and its purity was checked by performing SDS–PAGE using 15% polyacrylamide gel containing 0.1% SDS (Laemmli, 1970). The protein gel was stained with CBB R250.
Enzymatic assay of bmPSP
The bmPSP activity was assayed spectrophotometrically by measuring the formation of inorganic phosphate at 655 nm. The assay mixture consisted of 10 mM HEPES buffer (pH 7.0), 5 mM MgCl2, 1 mM L-phosphoserine, and approximately 0.99 mg/ml of bmPSP in a volume 250 μl. The reaction mixture was incubated at 37 °C for 30 min, and the reaction was
34
terminated by the addition of 250 μl 1 N HCl. After centrifugation at 13,000 rpm for 10 min, the supernatant was used for measuring the kinetics parameter towards the substrate L- phosphoserine (0.1–0.5 mM) by using theMichaelis-Menten equation. The optimum pH for wild-type bmPSP was determined by testing various pH values of the following buffers at the final concentration of 10 mM: glycine (pH 2.0 and 3.0); sodium acetate (pH 4.0 and 5.0);
MES (pH 6.0); HEPES (pH 7.0); Tris (pH 8.0 and 9.0). The optimum temperature for wild- type PSP was determined at different temperatures.
Determination of the concentration of inorganic phosphate
The concentration of inorganic phosphate was determined colorimetrically in 96-well polystyrene plates using the method of Fisher and Higgins (Fisher et al.,1994). Aliquots (80 μl) of the terminated incubation supernatant were mixed with (20 μl) malachite green reagent.
The plate was allowed to incubate for 30 min at room temperature, and the absorbance at 655 nm was determined using a microtiter plate spectrophotometer. One unit of activity was calculated as the amount of enzyme producing 1 μmol of inorganic phosphate/min.
Site-directed mutagenesis
The bmPSP mutants were produced through the chemical alteration of amino acids by employing the Quick-Change Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA), according to the previously described protocol (Yamamoto et al., 2017).
Mutagenesis was performed by using the following primers: 5′-
CAGCGGCCTGGTGCCGCGCGGCAGCCATATGGCTCTTTACAGTTTGAAAAGCA ACCT-3′ (sense) and 5′-
35
TGCCTTCATCTTGTATGACGGTCGAGTCTACAGCGAAGCAAACGCAGTCCGCTG TCCT-3′ (antisense) for D45A, 5′-
TCGACCGTCATACAAGATGAAGGCATCGATGAACTGGCCAAGTTCTGCGGGAA AGG-3′ (sense) and 5′-
TGCCTTCATCTTGTATGACGGTCGAAGCTACATCGAAGCAAACGCAGTCCGCTG TCCT-3′ (antisense) for D47A, 5′-
ATGAAGCCGTCAGCAGGTGGGCTAGCCTCGGCATCGGTAGCCCCAGCCCCGAC GATCACCAGGCGCTGGTACC-3′ (sense) and 5′-
TTCCTTTCGGGCTTTGTTAGCAGCCGGATCCTTACCAGGCTGCCTTCTTCTTGAC TTCGTCACGCACCACGTTACCCCCGAAGCCAATGAAGCCGTCAGCAGGTGGGC TA-3′ (antisense) for D204A. The plasmid harboring the bmPSP cDNA was used as a template for PCR. The whole length of the altered cDNA was verified by DNA sequencing using the 3730xl DNA Analyzer (Applied Biosystems,Foster City, CA, USA).
36 3.3. RESULTS
3.3.1. Cloning and sequencing of the bmPSP cDNA
The cDNA encoding 236 amino acids (Fig.3.1) with a calculated Mw and pI 26,150 and 6.21, respectively. The assumed amino acid sequence of bmPSP shared 60%, 59%, 59%, 20%, and 14% identity with the amino acid sequences of Bos taurus PSP (BtPSP), Homo sapiens PSP (HsPSP), Rattus norvegicus PSP (RnPSP), Mycobacterium tuberculosis PSP (MtPSP), and Hydrogenobacter thermophiles PSP (HtPSP) (Fig.3.1). Like other PSPs, all important amino acid residues involved in the catalytic site of bmPSP were conserved (Fig. 3.1). A phylogenetic tree analysis revealed that bmPSP was evolutionary related to the insect PSP (Fig. 3.2).
3.3.2. Localization of the bmPSP mRNA in various tissues
The relative level of the bmPSP mRNA in different tissues of B. mori was examined through qPCR. It was found that the bmPSP mRNA was present in all tested tissues of silkworm.
Higher bmPSP mRNA level was observed in the fat body (76% more that in the silk gland) and mid-gut (43% more than that in the silk gland) of silkworm compared to its expression in the silk gland. The lower and lowest amounts were observed in the testis (45% less than that in the silk gland), ovary (54% less than that in the silk gland) and hemocyte (88% less than that in the silk gland) respectively (Fig. 3.3.A). I also examined the level of mRNA of bmPSP in the MSG of the WT and variant b94 silkworms and found that the MSG of the
37
silkworm b94 contained very low (9-fold lesser than that of the MSG of the WT silkworm) amount of bmPSP mRNA than the MSG of the WT silkworm (Fig. 3.3.B).
3.3.3. Functional characterization of bmPSP
bmPSP was overproduced with an approximate molecular mass of 27,000 in E. coli Rosetta (DE3) pLysS cells (Fig. 3.4.A). The optimal results for the purification of bmSHMT by a nickel column and gel filtration would show only one band of protein at 52,000 in the elution fraction. (Fig. 3.4.B). It displayed the highest activity at pH 7.0 (Fig. 3.5.A) and at 37 °C (Fig. 3.5.C). More than 75% of the highest activity was achieved at pH 6.0–7.0 (Fig. 5.5.B) and at temperatures ranging from 30 to 40 °C (Fig.3.5. D). Like other PSPs, bmPSP was also dependent on divalent cations for its catalytic function. I examined the effect of Mg2+, Mn2+, and Ca2+ on bmPSP activity and found that bmPSP shows the highest activity in the presence of Mg2+ cations, and the activity was lost in the absence of divalent cations. This enzyme lost approximately 63% and 80% of activity, when Mn2+ and Ca2+ were used in place of Mg2+
ions, respectively (Table 3.2). The enzyme activity was appraised at optimal temperature and pH, indicating that the Km, Vmax, andkcat/Km values for L-phosphoserine were 5.2 × 10-2 (±
4.5 × 10-3) mM, 9.48 × 10-4 (± 1.0 × 10-5) units mg-1, and 7.9 × 10-3 mM-1S-1,respectively (Table 3.1).
3.3.4. Site-directed mutagenesis
A previous study reported that in human PSP, the residues D20, D22, and D179 are important for enzymatic hydrolysis (Peeraer et al., 2004). In bmPSP, the corresponding residues are D45, D47, and D204. For probing the role of amino acid residues in bmPSP activity, we
38
constructed three mutants named D45A, D47A, and D204A. All mutants were successfully overexpressed and purified to homogeneity. By determining the kinetic parameters of all three mutants for L-phosphoserine, we found that the replacement of the highly conserved amino acids by Ala resulted in the reduction of phosphoserine phosphatase activity. The catalytic efficiencies (kcat/Km) of D45A, D204A, and D47A decreased by 9-fold, 15-fold, and 38-fold of the catalytic efficiency of wild-type bmPSP, respectively (Table 3.1), indicating that these residues are important for bmPSP activity. Besides the catalytic activities, these Asp residues (D45, D47, and D204) play important roles in structural stability by forming a carboxylate through the generation of excess negative charge in the binding pocket.
39
3.4. DISCUSSION
In the present study, I identified and characterized a Mg2+-dependent phosphoserine phosphatase that is involved in the dephosphorylation of L-phosphoserine to produce L-serine in B. mori. The bmPSP cDNA consisted of 236 amino acid residues and showed a theoretical molecular mass of 26,150. The PSPs from humans (25,000) and Hydrogenobacter thermophilus (25,000) (Collet et al., 1997; Chiba et al., 2012) have similar molecular weights, whereas a higher molecular mass was observed for the PSPs isolated from the bovine brain (65,000) and Arabidopsis thaliana (47.349) (Antoine et al., 1974; Ho et al., 1999). The multiple sequence alignment results revealed that the identified bmPSP protein shared homology with HsPSP, RnPSP, HtPSP, and MtPSP, and this enzyme conserved all amino acid residues crucial for its catalytic function and structural stability (Fig 3.1). A phylogenetic analysis confirmed that bmPSP was evolutionary related to the insect PSP (Fig.
3.2). A qPCR analysis exhibited that the bmPSP mRNA was existed in all tested tissues (Fig.
3.3.A). The bmPSP mRNA expression in the silk gland indicates that this enzyme plays an important role in silk formation by the biosynthesis of L-serine, which is the major amino acid constituent of sericin. It has been reported previously that L-serine acts as a basic structural unit for the production of important cell constituents, including glycine, cysteine, methionine, serine, purines, thymidine, and neuromodulator D-serine in mammal (Snell, 1984; Snyder & Kim, 2000). Therefore, the existence of bmPSP mRNA in other tissues of silkworm indicate that this enzyme plays an important role in the synthesis of above- mentioned cell metabolites in silkworm. The analysis of the bmPSP mRNA level between the MSGs of the wild-type silkworm (p50T) and a sericin-deficient variant b94, it was
40
observed that the level of bmPSP mRNA was very low (9-fold lower) in the b94 strain compared to the wild-type p50T strain (Fig. 3.3.B). I also found that the MSG of the b94 variant contained lower amount of the bmPSAT mRNA compared to the p50T strain (chapter IV). Together, these results show that the relatively lower amount of the bmPSP mRNA in the MSG of the b94 variant might be one of the rate-limiting factors for sericin production in this variant.
The optimum pH required for the optimal activity of bmPSP was 7.0 (Fig. 3.5.A).
The PSPs isolated from the human brain (pH 6.6) (Veeranna & Shetty, 1990) and Methanococcus jannaschii (pH 7.5) (Wang et al., 2002) showed their maximal activity at almost neutral pH, whereas those isolated from mouse (pH 5.0) (William, 1967) and bovine brain (pH 5.8–6.2) (Antoine et al., 1974) showed their activity at a pH lower than that required for the maximal activity of bmPSP. The maximal enzymatic activity of bmPSP was achieved at 37 °C (Fig. 3.5.C), and more than 80% of the enzymatic activity was achieved at temperatures ranging from 30 to 40 °C (Fig. 3.5.D). The maximal activity of the PSPs isolated from M. jannaschii (Wang et al., 2002) and Thermococcus onnurineus (Jung et al., 2013) was observed at a temperature higher than that required for the maximal activity of bmPSP. I have exemplified the biochemical and functional confirmation of the assertion that PSP plays a crucial role in serine synthesis in B. mori. The kinetics results proposed that bmPSP was able to recognize L-phosphoserine as a substrate. Increasing the Km value (5.2 × 10-2 mM) of the substrate L-phosphoserine from the reported Km values of the PSPs isolated from human brain (3.6 x 10-2 mM) (Veeranna & Shetty, 1990), rat brain (0.160 mM) (Hawkinson et al., 1996), Hydrogenobacter thermophilus (1.6 mM) (Kim et al., 2016),M.
jannaschii (0.62 mM) (Wang et al., 2002), and Mycobacterium tuberculosis (0.38 mM)
41
(Gregory, 2017) revealed that bmPSP had lower substrate affinity than the PSPs of the above- mentioned organisms. However, the substrate affinity of bmPSP was higher than that of the PSP isolated from T. onnurineus (8.1 x 10-2 mM) (Jung et al., 2013). The calculated catalytic efficiency (kcat/Km) of bmPSP was lower than that of the PSP isolated from other organisms.
Collet et al. (1999) reported that in human PSP, the substitution of Asp20, Asp22, and Asp179 (in bmPSP Asp45, Asp47, and Asp204) residues by Ala promoted the reduction of enzymatic activity. Asp20 mediates a nucleophilic attack on the phosphate of the substrate
L-phosphoserine, thereby contributing to the breakdown of phosphoserine to release serine and phosphoaspartyl (Asp20) intermediate. Asp22 acts as an acid that donates a proton to serine, thus facilitating the expulsion of the leaving group. Furthermore, this residue activates the water molecule present in the vacant site by extracting a proton, which causes a nucleophilic attack on the phosphoaspartyl intermediate, thereby facilitating the release of the enzyme and separation of inorganic phosphate. The mutation of Asp45 and Asp47 results in a 9- and 15-fold decrease in the hydrolytic activity of the wild-type enzyme, respectively, suggested that these residues may be crucial for binding to the phosphate group of the substrate phosphoserine in bmPSP. In human PSP, Asp179 (Asp204 in bmPSP) has the affinity for binding to Mg2+ ions (Collet et al., 1999). The mutational studies of Asp204 indicate that this residue mediates binding to the divalent cation Mg2+.
42 Acknowledgements
I am thankful for Dr. Kohji Yamamto to excise fat body, clone, and sequence bmPSP, and for Aiko Hirowatari to prepare expression vector for bmPSP.
43
Fig. 3.1. Multiple alignment of the deduced amino acid sequence of PSP.
Sequence alignment was prepared using the Clustal Omega program. The PSP sequences of various organisms were collected from the NCBI and Uni-Prot databases: bmPSP (determined in the study), Bos taurus (BtPSP; NP_001039820.1), Homo sapiens (HsPSP;
P78330), Rattus norvegicus (RnPSP; NP_001009679.1), Hydrogenobacter thermophiles (HtPSP; D3DFP8), and Mycobacterium tuberculosis (MtPSP; P9WGJ3). Red and black boxes indicate the L-phosphoserine and inorganic phosphate (Pi) binding sites, respectively.
The shaded residue indicates the Mg2+ binding site.
44
Fig. 3.2. Phylogenetic analysis of PSP amino acid sequences.
The phylogenetic tree was prepared with the neighbor-joining software using the amino acid sequences of different PSPs gathered from the NCBI and Uni-Prot databases. The species name and accession number are depicted within the figure. The arrow represents the phylogenetic position of the bmPSP. Hydrogenobacter thermophilus (D3DFP8) was used as outgroup. Scale bar (0.1) shows number of amino acid substitutions per site.
Insects
45
Fig. 3.3. Localization of the bmPSP mRNA
(A) Distribution of the bmPSP mRNA in various tissues (B) Comparison of bmPSP mRNA between the middle silk glands of the wild-type and variant b94 silkworms. We performed a qPCR to detect the bmPSP mRNA in different tissues. The amounts of mRNA expressed in different tissues were analyzed by qPCR as described in the Materials and Methods. The error bars represent the standard errors obtained from three experiments. W and MSG indicate wild-type and middle silk gland, respectively.
(A)
(B)
46
Fig. 3.4. (A) Overexpression and (B) Purification.
(A) For overexpression of bmPSP, 1 mM IPTG was used as an inducer. Lane 1 represents the protein molecular size marker. Lane 2 shows the bmPSP protein band without induction with 1 mM IPTG, whereas lane 3 shows the band corresponding to the bmPSP protein overexpressed with 1 mM IPTG. The arrow indicates the overproduced bmPSP protein, and (B) Purification of bmPSP was done as described in the Materials and Methods section. Lanes 1 and 2 are the fractions that eluted from Ni2+- resin and protein molecular marker, respectively. The arrow indicates the band of purified bmPSP protein.
(A) (B)
47
Fig. 3. 5. (A) Optimum pH and (B) pH stability
(A) Enzyme activity was assayed at 37 °C and pH 2.0–9.0, and (B) pH stability was determined after pre-incubating bmPSP at various pH values and 4 °C for 24 h, and its net activity was measured at 37 °C and at pH 7.0. The maximal value obtained was set to 100%.
(B) (A)
48
Fig. 3. 5. (C) Optimum temperature and (D) Thermostability.
(C) The net activity was determined by using Hepes buffer with pH 7.0 at 20 - 45 °C, and (D) For determining thermostability, the enzyme solution was pre-incubated at various temperatures for 30 min before the relative activity was assayed.
(C)
(D)
49
Table 3. 1. Kinetic properties of bmPSP towards L-Phosphoserine. The Michaelis-Menten
equation was used for calculating the kinetic parameters
** One unit of enzyme activity was defined as the amount of enzyme producing 1 μmol of inorganic phosphate/min.
Protein Km
(mM)
Vmax
(units mg-1)
kcat/Km (mM-1S-1) Wild-type
PSP 5.2 × 10-2 (± 4.5 × 10-3) 9.48 × 10-4 (± 1.0 × 10-5) 7.9 × 10-3 D45A 2.8 × 10-1 (± 6.0 × 10-2) 5.75 × 10-4 (± 3.0 × 10-5) 8.8 × 10-4 D47A 0.34 × 10-1 (± 4.1 × 10-1) 1.66 × 10-3 (± 1.2 × 10-4) 2.1 × 10-4 D204A 7.3 × 10-1 (± 7.8 × 10-2) 8.97 × 10-4(± 3.8 × 10-5) 5.4 × 10-4
50
Table 3. 2. Effect of various divalent cations on bmPSP activity Supplement (5 mM) Relative activity (%)
MgCl2 100 ± 0.004
MnCl2 36.69 ± 0.003
CaCl2 19.34 ± 0.002
51
Chapter IV
Identification and biochemical characterization of the serine hydroxymethyltransferase from B. mori
4.1. INTRODUCTION
Serine hydroxymethyltransferase (SHMT; EC 2.1.2.1) found as a part of 1C metabolism, catalyzes the relocation of the hydroxymethyl group of L-serine to THF to produce glycine and N5, N10-MTHF by using pyridoxal-5-phosphate (PLP) as a cofactor. MTHF acts as the main 1C donor in the 1C metabolic reaction for the production of serine, thymidylate, purines, and methionine (Schirch, 1982; Jala et al., 2002). The SHMT-dependent reversal catalytic reaction of glycine and THF is one of the most upcoming ways to yield L-serine (Fig. 1) (Hamilton et al., 1985; Hsiao & Wei, 1985). The enzyme SHMT is distributed in prokaryotic and eukaryotic cells. In mammals, SHMT has been shown to exist in two forms viz. cytosolic (SHMT1) and mitochondrial (SHMT2) (Schirch, 1982; Stover et al., 1997; Girgis et al., 1998). However, only cytosolic form is present in the parasite Trypanosoma cruzi (Capelluto et al., 2000). The produced 1C THF pools are interconverted by the mono- or bi- or tri- functional MTHFD. To identify 1C metabolism in silkworm, a Lepidopteran model and biologically important insect, as described in chapter V, I have characterized another important 1C metabolic bi-functional MTHFD from this insect. In terms of the catalytic and functional studies of SHMT from various sources, it is enticing to identify and investigate the properties of this enzyme facilitate to explore knowledge 1C metabolism in silkworm. In
52
present study, I dispatched the identification, distribution of mRNA and characterization of 1C metabolic SHMT from B. mori (bmSHMT).
53
4.2. MATERIALS AND METHODS
Insects
B. mori larvae (p50T strain) were nurtured on mulberry leaves at the Institute of Genetic Resources, Kyushu University (Fukuoka, Japan). The fat body was excised from the day-3 fifth-instar larvae in ice-cold 0.75% NaCl, rapidly frozen in liquid N2, and saved at −80 °C prior to use.
Molecular cloning and sequencing of the bmSHMT cDNA
Total RNA was derived from the fat body of B. mori larvae using Sepasol-RNA 1 (Nacalai Tesque), based on the manufacturer’s protocol. The cDNA was produced using the SuperScript II First-Strand Synthesis System for PCR F (Thermo Fisher Scientific, Carlsbad, CA) and an oligo-dT primer. Subsequently, the cDNA was amplified by PCR using the
following two oligonucleotide primers 5′-
TAGTTCCCATATGAGCGCCAAGTTACTAAA-3′ (sense) and 5′-
TTGGATCCTTAATAT TTATCGAACCA GGC-3′ (antisense). The primers were designed based on a sequence found from the SilkBase EST database (http://silkbase.ab.a.u- tokyo.ac.jp/cgi-bin/index.cgi). The underlined and double-underlined portions in the primer sequence indicate Ndel and BamHI restriction enzyme sites, respectively. The following PCR program was used: one cycle of initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 59 °C for 1 min, and extension at 72 °C for 2 min; followed by one cycle of extension at 72 °C for 10 min. After ligation with the pGEM-T Easy Vector (Promega, Madison, WI), the produced bmSHMT cDNA was
54
inoculated to E. coli DH5α cells. The NCBI and Uni-Prot databases were used to get the amino acid sequence of SHMTs, and Clustal Omega were used for sequence alignment preparation. A phylogenetic tree was made by NJ plot software.
qPCR analysis
A qPCR was performed to confirm the presence of the mRNA expression profiles in bmSHMT. The SuperScript II First-Strand Synthesis System was used for synthesizing cDNAs from mRNAs. The housekeeping gene Bmrp49 was used as control for cDNA normalization. Primer fixes for bmSHMT and B. mori ribosomal protein 49 (Bmrp49) were sequenced. bmSHMT F, (forward) 5′-AGCGTGGCCTGCAACAAGAAC-3′; bmSHMT R, (reverse) 5′-TCTTGTGGTCAACGCTGGTGTAC-3′; Bmrp49 F, (forward) 5′- GATGTGTTTTATATTC-3′: Bmrp49 R, (reverse)
5′-GCATCATCAAGATTTCCAGCTC-3′ were used as primer sequences. The qPCR was performed in a total volume 20 μl containing 10 μl SYBR® Green reagents, 1 μl (100 ng/μl) cDNA, 1.6 μl (800 nM) each of forward and reverse primers and 5.8 μl distilled water. qPCR magnification initiated with a denaturation step at 95 °C and then 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min, and extension at 60 °C for 1 min. Expression level was standardized against alike Bmrp49 levels and set as the bmSHMT/Bmrp49 ratio.
.
Overexpression and purification of bmSHMT and mutants
The bmSHMT clone was digested with NdeI and BamHI, ligated into the expression vector pET‐15b (Merck Millipore, Darmstadt, Germany), and transformed into competent E. coli BL21 (DE3) cells (Merck Millipore). The cells were then cultured at 37 °C in 250 mL. The
55
LB medium containing 250 µl ampicillin (50 mg/ml) until OD600 become approximately 0.50.
Thereafter, the culture was kept on ice for 30 min. After addition of 300 µl of 10 mM IPTG as an inducer it was cultured overnight at 20 °C. The cells were harvested through centrifugation and stored at –20 °C until use. The cell pellet was resuspended with 20 mM Tris-HCl with 0.5 M NaCl; pH 8.5, containing 0.1 mg/mL lysozyme and 10 μM PLP. After incubating at 37 °C, cells were agitated by sonication. The refined supernatant was subjected to Ni2+-affinity chromatography (His-Pur Ni-NTA resin, Thermo Scientific, Meridian Rd., Rockford, U.S.A). After washing with the 20 mM Tris-HCl with 0.5 M NaCl (pH 8.5) buffer, 20 mM Tris-HCl with 0.5 M NaCl; pH 8.5 with 0.2 M imidazole was used in for eluting the protein. The purity of the enzyme was checked by performing SDS-PAGE using 15%
polyacrylamide gel containing 0.1% SDS (Laemmli, 1970). The protein gel was stained with CBB R250.
Site-directed mutagenesis
The bmSHMT mutants were produced through the chemical alteration of amino acids by employing the Quick-Change Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA), based on the previously described protocol (Yamamoto et al., 2017). Mutagenesis was performed by using the primers: 5′-CCGTTTATACCGGCATTG
TCGAACCCGCTGGCAGGATAATGGGGTTAGATTTACC-3′ (sense) and 5′- GGTAAATCT
AACCCCATTATCCTGCCAGCGGGTTCGACAATGCCGGTATAAACGG-3′
(antisense) for H119A, 5′-
TGGGGTTAGATTTACCTGACGGTGGAGCTCTCACCCATG
56 GTTTCTTTACTGCTAC-3′ (sense) and 5′-
GTAGCAGTAAAGAAACCATGGGTGAGAGCTCCACCGTCAGGTAAATCTAACCC CA-3′ (antisense) for H132A, 5′-
ATTTACCTGACGGTGGACATCTCACCGCTGGTTTCTTTACTGCTACTAAAAAAA TATCTGCTAC-3′ (sense) and 5′-
GTAGCAGATATTTTTTTAGTAGCAGTAAAGAAACCAGCGGTGAGATGTCCACC GTCAGGTAAAT-3′ (antisense) for H135A. The plasmid harboring the bmSHMT cDNA was used as a template. The whole length of the altered cDNA was verified by DNA sequencing using the 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Enzymatic assay of bmSHMT and mutants
The bmSHMT activity was assayed indirectly through coupling its reaction with the reaction of bmMTHFD. bmSHMT catalyzed the alteration of serine and THF to glycine and MTHF (CH2 -THF) by utilizing PLP as a cofactor (1) and then, bmMTHFD used 5,10-CH2-THF and NADP+ as a substrate to yield 5,10-methenyl-THF (CH+-THF) and NADPH (2). The reaction advancement was monitored by the increase of NADPH with the Nano Drop® ND- 1000 spectrophotometer at 340 nm.
(1)
(2)
Glycine 5,10-CH2 -THF THF
L-Serine bmSHMT
NADP+ 5,10-CH2 -THF
NADPH
bmMTHFD
PLP
5,10-CH+ -THF
57
The assay mixture was contained Britton-Robinson buffer; pH 3.0, 2 mM DTT, 1 mM EDTA, 240 µM NADP+, 4 mM L-serine, 0.044 mM THF, 1.5 µl MTHFD (10 mg/ml) and 2 µl SHMT (8 mg/ml) in a volume of 50 μl. The kinetic constants of bmSHMT (WT and mutants) were determined for THF and serine. For THF, THF concentrations were varied from 0.0448 to 0.2688 mM while fixing the serine concentration at 4 mM. For serine, in reaction mixture the concentrations of serine were varied from 4 to 24 mM, fixing the THF concentration at 0.0448 mM. After incubation at 30 °C for 10 min, reaction advancement was recorded. The Km and Vmax values for THF and serine were determined by the Line-weaver Burk Plot of initial velocity versus substrate concentration.
58 4.3. RESULTS
4.3.1. Cloning and sequencing of cDNA encoding bmSHMT
The bmSHMT cDNA was obtained by RT-PCR using total RNA from B. mori fat body. The cDNA encoding 465 amino acids (Fig. 4.1) with a computed Mw of 51,220 and the pI of bmSHMT calculated from the assumed amino acid residues, was 7.56. The assumed amino acid sequence of this presumptive bmSHMT shared 71%, 64%, 63%,63%, 60%, and 59%
identities to Drosophila melanogaster (common fruit fly) SHMT (dSHMT), human cytosolic SHMT (hcSHMT1), rabbit cytosolic SHMT (rcSHMT1), sheep liver cytosolic SHMT (scSHMT), human mitochondrial SHMT (hmSHMT2), and rabbit mitochondrial SHMT (rmSHMT2), respectively (Fig. 4.1). The bmSHMT sequence is almost equally identical to both cytosolic (SHMT1) and mitochondrial (SHMT2) sequence. Furthermore, bmSHMT conserved amino acid residues, which are important for the enzymatic activity of SHMTs. A phylogenetic tree analysis showed that the present bmSHMT was found to be evolutionarily close to the SHMT of insect PSP (Fig.4.2).
4.3.2. Expression of bmSHMT mRNA
To investigate the expression level of mRNA, total RNAs of fat body, hemocyte, ovary, silk gland, and midgut of day 3 fifth-instar larvae of silkworm were reverse transcribed to cDNA by RT-PCR and used as templates. These results showed that the bmSHMT mRNA was present in all tested tissues (Fig.4.3. A). The bmSHMT mRNA expression is higher in the fat body; lower in the testis (40% less than fat body), ovary (47% less than fat body), hemocyte, and silk gland (78% less than fat body); and the lowest in the mid-gut (96% less than fat body). The analysis of the bmSHMT mRNA between the middle silk gland (MSG) of the
59
standard silkworm (p50T strain) and sericin-deficient b94 strain showed a reduction of approximately 60% expression in the b94 strain (Fig. 4.3.B)
4.3.3. Overexpression and purification of bmSHMT and mutants
bmSHMT and mutants (H119A, H132A, and H135A) were overproduced in E. coli. The overexpressed enzymes showed a band of approximately at 52,000 (Fig. 4.4.A), which is also consistent with the predicted molecular mass (51,220). Recombinant bmSHMT was purified to homogeneity by affinity chromatography (Fig. 4.4. B). The optimal results for the purification of bmSHMT by a nickel column would show only one band of protein in the elution fraction. When analyzed by SDS-PAGE, the purified bmSHMT protein showed a single band.
4.3.4. Biochemical and functional characterization of bmSHMT
The biochemical characteristics of bmSHMT were measured by applying NADP+ and THF as a cofactor and substrate, respectively. The supreme activity of bmSHMT was found at a temperature at 30 °C (Fig. 4.5.A) and more than 75 % of the capital activity was obtained between 20 to 40 °C (Fig. 4.5.B), the bmSHMT activity eliminated rapidly at a temperature
>40 °C. The optimum pH for bmSHMT was 3.0 in this study (Fig. 4.5.C), and this was stable at pH 3.0–6.0, and over 65% of the activity was obtained between pH 3.0 and 6.0. Moreover, bmSHMT was responsive to high pH and nearly no activity over pH 8.0 (Fig. 4.5.D).
Therefore, bmSHMT was stable under acidic condition. The enzymatic activity was deliberated at the standard reaction system, indicating that the Km and kcat/Km values of THF