九州大学学術情報リポジトリ
Kyushu University Institutional Repository
リグニン初期熱分解と二次気相反応の詳細化学反応 速度モデリング
古谷, 優樹
https://doi.org/10.15017/1931946
出版情報:Kyushu University, 2017, 博士(工学), 課程博士 バージョン:
権利関係:
I
Detailed Chemical Kinetic Modeling of Primary and Secondary Pyrolyses of Lignin
(リグニン初期熱分解と二次気相反応の 詳細化学反応速度モデリング)
by
Yuki Furutani (2018)
Department of Applied Science for Electronics and Materials, Interdisciplinary Graduate School of Engineering Sciences,
Kyushu University, Japan
I
Contents
Chapter 1: General Introduction---1
1.1. Background---1
1.2. Overview of Biomass Thermochemical Conversion---2
1.3. Detailed Chemical Kinetic Models---2
1.3.1. Benzene Oxidation---3
1.3.2. Phenol Pyrolysis---4
References---8
Chapter 2: Predicting Molecular Composition of Primary Product Derived From Fast Pyrolysis of Lignin with Semi-Detailed Kinetic Model---13
2.1. Introduction---13
2.2. Methods and Modeling---14
2.2.1. Virtual Compounds for Approximating Lignin Structures---14
2.2.2. Estimation of Heating Rate---15
2.2.3. Detailed Chemical Kinetic Model for Lignin Pyrolysis---16
2.3. Results and Discussion---17
2.4. Conclusions---18
References---19
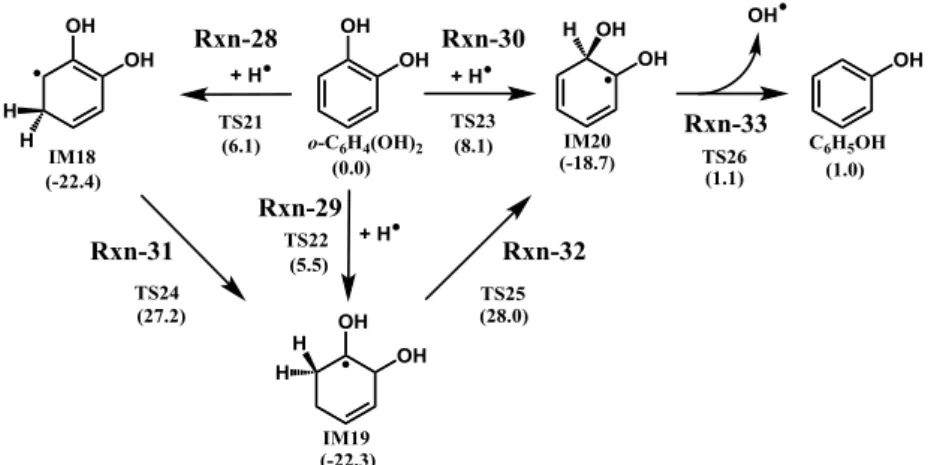
Chapter 3: Theoretical Study on Reaction Pathways Leading to CO and CO2 in the Pyrolysis of Resorcinol---32
3.1. Introduction---32
3.2. Computational Method---33
3.3. Results and Discussion---34
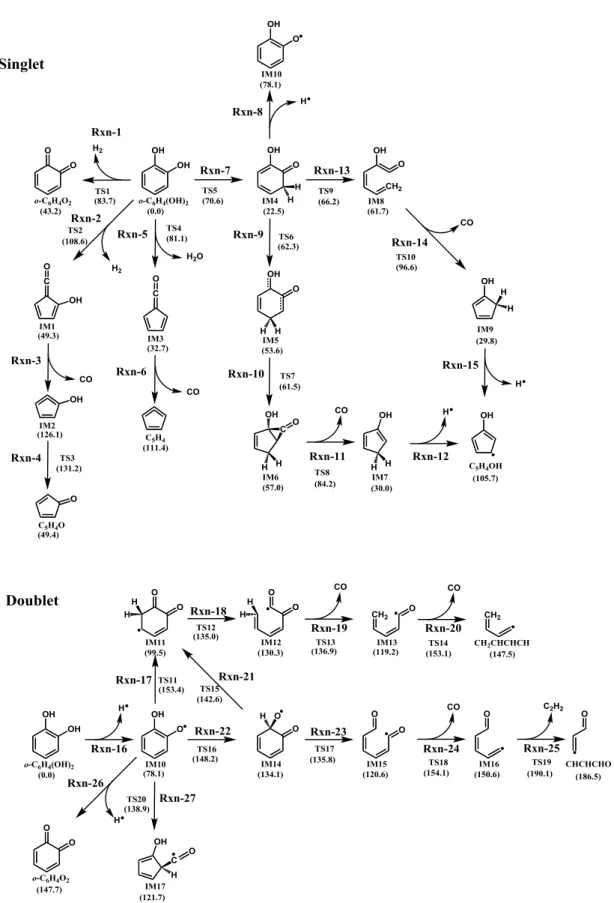
3.3.1. Proposal of Reaction Pathways for Resorcinol Pyrolysis---34
3.3.2. Optimized Resorcinol Configurations---35
3.3.3. Potential Energy Surfaces in the Pyrolysis Process for Each Reaction Pathway--35
3.3.4. High-Pressure Limiting Rate Constants---36
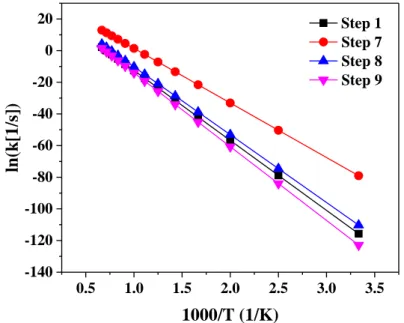
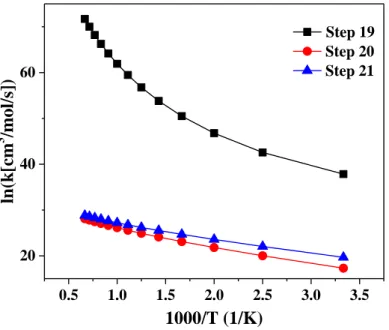
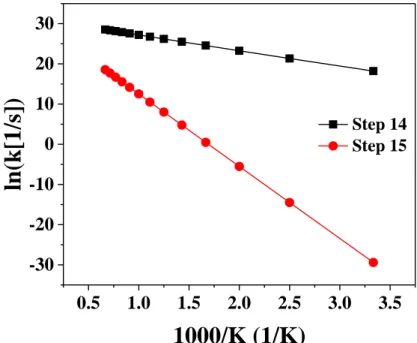
3.3.4.1. Rate Constants for Step 1, 7, 8, and 9---36
3.3.4.2. Rate Constants for Steps 2 and 8---36
3.3.4.3. Rate Constants for Steps 14 and 15---37
3.4. Conclusions---37
References---37
II
Chapter 4: Theoretical Study on the Kinetics of Thermal Decomposition of Guaiacol
and Catechol---49
4.1. Introduction---49
4.2. Computational Method---50
4.3. Results and Discussion---51
4.3.1. Proposal of Pyrolysis Pathways on Potential Energy Surfaces---51
4.3.2. High-Pressure Limiting Rate Constants Calculated with Transition State Theory- ---53
4.4. Conclusions---54
References---54
Chapter 5: Theoretical Study on Elementary Reaction Steps in Thermal Decomposition Processes of Syringol-Type Monolignol Compounds---70
5.1. Introduction---70
5.2. Computational Method---71
5.3. Results and Discussion---71
5.3.1. Reaction Pathways and Kinetic Database on HDPP Pyrolysis---72
5.3.2. Reaction Pathways and Kinetic Database for the Pyrolysis of HHDPP, Sinapyl alcohol, and HDPPD---73
5.3.3. Reaction Pathways and Kinetic Database of Syringol Pyrolysis---73
5.3.4. Reaction Pathways and Kinetic Database on Pyrogallol Pyrolysis---74
5.4. Conclusions---75
References---75
Chapter 6: Theoretical Study on Elementary Reaction Steps in Thermal Decomposition Processes of Phenol-Type Monolignol Compounds---90
6.1. Introduction---90
6.2. Computational Method---91
6.3. Results and Discussion---91
6.3.1. Thermal Decomposition Pathways and Kinetic Parameters for HPP Pyrolysis-92 6.3.2. Thermal Decomposition Pathways and Kinetic Parameters on Pyrolysis of HHPP, p-Coumaryl Alcohol, and HPPD---93
6.4. Conclusions---94
References---94
III
Chapter 7: Coupling Detailed Chemical Kinetic Models of Primary and Secondary
Pyrolysis of Lignin---108
7.1. Introduction---108
7.2. Calculation Method---109
7.2.1. Reaction Mechanism---109
7.2.2. Numerical Simulation---110
7.3. Results---110
7.4. Conclusions---111
References---111
Chapter 8: General Conclusions---118
Acknowledgements---120
Appendix A---121
Appendix B---141
Appendix C---145
Appendix D---147
1
Chapter 1: General Introduction
1.1. Background
Ever since the industrial revolution, humanity has consumed a huge amount of energy;
thus, the global energy demand in 2040 is predicted to increase 37% higher than current level [1]. Currently, fossil fuel energy consisting of coal, petroleum, and natural gas made up 78.4% in the total world energy consumption in 2013 [1]. However, this massive fossil fuel energy consumption has caused various issues such as climate change, greenhouse gas emissions and the depletion problem of fossil fuels. As a result, the interest in renewable energy is increasing, and in particular biomass utilization has received broad attention due to the only renewable energy source that can be converted into liquid fuel and utilized as feedstock in chemicals production [2]. Current biomass share in world’s total primary energy consumption is approximately 10%, and this will be increased in developing countries during the next 30 years [1]. Biomass is recognized as a CO2 neutral resource because the amount of carbon released back into the atmosphere is equivalent to that converted into new plants through photosynthesis during its lifetime. Boman et al.
[3] demonstrated that the CO2 emissions for the full fuel cycles could be decreased by more than 90% if biomass resources were utilized not only as power supply but also as external input fuels required for the production and transportation of the biomass feedstock. Moreover, the lower emission of environmentally-harmful gases, such as sulfur oxide (SOx) and nitrogen oxide (NOx), leads to the reduction global acid rain formation [4].
In addition to the environmental and energy security aspects, biomass utilization could contribute to job creation in local community, enhancement of competitiveness in industries, and the activation of agriculture, forestry, and fishery in rural areas because biomass energy is recognized as a “local production for local consumption”. In Japan, therefore, “Biomass Nippon Strategy” was approved to promote both the biomass energy and material utilization in December 2002 [5]. The goal of this strategy was to expand biomass utilization and develop the relating technologies. The strategy mentions the four reasons to promote biomass utilization: (1) creation of a regional employment, (2) activation of forestry, fishery, and agriculture industries, (3) reduction of greenhouse gas emission, and (4) fostering and promotion of venture companies. Since then, a number of projects have been conducted both nationally and locally. For example, a centralized biomass treatment facility (biogas plant and composting plant) was built in Shikaoi town in Hokkaido in September 2007 [6]. Sumitomo Corporation will start to construct the 50
2
MW wood biomass power plant in Sakata City of Yamagata Prefecture around June 2016 and the plant is scheduled to be put into commercial operation in May 2018 [7].
1.2. Overview of Biomass Thermochemical Conversion
An advantage of biomass utilization is that biomass resources can be used in various types of energy application, including electric and heat generation for industrial facilities and homes. Biomass conversion technologies to such useful energies can be separated into four process technologies: direct combustion process technology, biochemical process technology, agrochemical process technology and thermochemical process technology [8]. The final energy products and the advantages of these process technologies were listed in Table 1. Direct combustion technology is the main process to convert biomass energy into heat, mechanical power and electricity. This system is available in the size range from a few MW up to more than 100 MW [9]. Biochemical processes, using fermentation, converts biomass into biological hydrogen gas and ethanol via acid/enzymatic hydrolysis. Biomass resources with high-moisture content, such as the herbaceous plant sugarcane, is more economically suited to biochemical process which is `wet/aqueous' conversion [10]. Agrochemical processes are the mechanical extraction method to produce bio-diesel energy form. For instance, bio-diesel is directly extracted by crushing seed crops containing a high percentage of oil [11,12]. Thermochemical processes are able to be subdivided into gasification and pyrolysis. Gasification converts biomass with a low moisture content (e.g. wood chips) into syngas products, such as hydrogen (H2), carbon monoxide (CO) and methane (CH4). The syngas is converted into both electricity and heat in a gas engine. Biomass gasification has been developed commercially owing to two main factors: a higher level of electricity and heat generation efficiency even in small-scale plant, and a lower level of exhaust emissions of major air pollutants. Pyrolysis is the thermochemical conversion process in the absence of air to produce a bio-oil, a charcoal and a gaseous.
1.3. Detailed Chemical Kinetic Models
Kinetic models are recognized to be effective in the prediction of the biomass thermochemical conversion and the suggestion for the improvement of the processes.
Lumped kinetic models are conventionally used to elucidate the thermochemical mechanism through grouping species and reaction [13]. However, this model is too simple to describe the gas composition at the molecular level. Detailed chemical kinetic model
3
(DCKM) established based on the elementary reaction steps is a powerful tool to predict reactor performances and design the optimized reactor at the molecular level. In order to develop the DCKM, the derivation of rate constants in each elementary reaction is indispensable. Recently, computational chemistry approaches using ab-initio calculation and transition state theory have been pointed out as one of the major fields relating to the high-accuracy theoretical estimation of the rate constants. In this section, firstly, the elementary reaction mechanisms and kinetics focusing on benzene oxidation and phenol pyrolysis are reviewed from the both aspects of experimental and theoretical approaches to verify the computational chemistry approaches.
1.3.1. Benzene Oxidation
The initial reactions of benzene oxidation involve the unimolecular decomposition and abstraction by H, O and OH radical pools to form phenyl radical (C6H5), phenoxyl radical (C6H5O) or phenol (C6H5OH) [14].
With the high concentration of an O radical in a combustion environment, the reaction of benzene with O atoms occurs as follows:
C6H6 + O ⇄ C6H5 + OH (Rxn-1)
C6H6 + O ⇄ C6H5O + H (Rxn-2)
C6H6 + O ⇄ C6H5OH (Rxn-3)
The investigation of the above overall reaction in the gas phase using mass spectrometry was performed by Ko et al. [15] Nguyen et al. [16] theoretically studied the potential energy surfaces (PES) of Rxn-1, Rxn-2, and Rxn-3 by using the complete basis set method (CBS-QB3), which is a series of high accuracy methods including a complete basis set extrapolation, and calculated the rate constants of overall reactions based on transition state theory (TST). The results obtained from the high-level quantum chemical calculation (obtained by Nguyen et al. [16]) were in good agreement with those from the experiment (obtained by Ko et al. [15]) as shown in Figure 1.
In the high OH radical concentration, furthermore, benzene reacts with an OH radical as follows:
C6H6 + OH ⇄ C6H5 + H2O (Rxn-4)
C6H6 + OH ⇄ C6H5O + H2 (Rxn-5)
C6H6 + OH ⇄ C6H5OH + H (Rxn-6)
4
Seta et al. [17] investigated the overall rate constant of Rxn-4, Rxn-5 and Rxn-6 in the both fields of experiment and theoretical calculation. The rate constant was both experimentally derived by a shock tube/pulsed laser-induced fluorescence imaging method and theoretically predicted by TST calculations based on CBS-QB3 method.
According to the comparison in Figure 2, the experimentally-obtained rate constants were closely consistent with the calculation results.
1.3.2. Phenol Pyrolysis
At higher temperatures, C6H5OH decomposes into cyclopentadiene (C5H6) and CO as follows:
C6H5OH ⇄ C5H6 + CO (Rxn-7)
Horn et al. [18] performed the high-temperature pyrolysis experiment of C6H5OH by using the shock-tube technique to obtain the rate constant of Rxn-7. Xu et al. [19]
theoretically investigated the reaction potential energy surface at the G2M//B3LYP/6- 311G(d,p) level of theory, and obtained the rate constants by using TST. The theoretical prediction of the rate constant (obtained by Xu et al.[19]) well reproduced the experimental values (obtained by Horn et al.[18]) as shown in Figure 3.
In conclusion, computational chemistry with ab-initio calculation and TST is capable of providing extremely accurate rate constants. In this thesis, computational chemistry was applied to the development of the comprehensive DCKM for predicting lignin pyrolysis processes.
The purpose of this thesis is to construct the DCKM predicting both primary and secondary pyrolysis of lignin based on the computational chemistry approaches. This thesis consists of eight chapters as follows:
Chapter 1: General Introduction
General introduction was described.
Chapter 2: Predicting Molecular Composition of Primary Product Derived From Fast Pyrolysis of Lignin with Semi-Detailed Kinetic Model
5
A numerical approach is presented for predicting the yields of char and volatile components obtained from fast pyrolysis of three types of lignin (enzymatic hydrolysis lignin, EHL; organic extracted lignin, OEL; and Klason lignin, KL) in a two-stage tubular reactor (TS-TR) at 773–1223 K. The heating rate of lignin particle in the TS-TR was estimated at 102–104 K/s by solving the heat transfer equation. The pyrolytic behavior of lignin and the formation of products in the temperature rising process were predicted using a semi-detailed kinetic model consisting of 93 species and 406 reactions, and the predicted yields of 8 primary products (i.e., char, tar, CO, CO2, H2O, CH3OH, C2H6, and C3H6) were compared with experimental data for the critical evaluation. For EHL, the predicted yields of char and H2O were in good agreement with the experimental results at all temperatures. However, the numerical simulation overestimated tar yield and underestimated CO yield at high temperature probably due to a lack of the kinetic model of the tar cracking reaction. The predicted yields of CH3OH, C2H6, and C3H6 were close to the experimental values at high temperature by adding the detailed chemical kinetic model of the secondary vapor-phase reaction. Moreover, the model reproduced the experimental observation that among the three types of lignin the char yield increased in the order of EHL < OEL < KL, whereas the tar yield decreased.
Chapter 3: Theoretical Study on Reaction Pathways Leading to CO and CO2 in the Pyrolysis of Resorcinol
Possible pathways for the pyrolysis of resorcinol with the formation of CO and CO2 as final products were proposed and evaluated using ab initio calculations. Our experimental study revealed that large quantities of CO2 are generated in the pyrolysis of 1,3- dihydroxybenzene (resorcinol), while the pyrolysis of the dihydroxybenzene isomers 1,2- dihydroxybenzene (catechol) and 1,4-dihydroxybenzene (hydroquinone) produces little CO2. The fate of oxygen atoms in catechol and hydroquinone was essentially the formation of CO. In the proposed pathways, the triplet ground state m-benzoquinone was generated initially from simultaneous cleavage of the two O−H bonds in resorcinol.
Subsequently, the direct cleavage of a C−C bond of the m-benzoquinone diradical yields 2-oxidanylcyclopenta-2,4-dien-1-yl-methanone, which can be converted via two channels: release of CO from the aldehyde radical group and combination of the ketone radical and carbon atom in the aldehyde radical group to form the 6- oxabicyclo[3.2.0]hepta-2,4-dien-7-one, resulting in the release of CO2. Potential energy
6
surfaces along the proposed reaction pathways were calculated employing the CBS-QB3 method, and the rate constants at the high-pressure limit were also evaluated based on transition-state theory to assess the feasibility of the proposed reaction pathways.
Chapter 4: Theoretical Study on the Kinetics of Thermal Decomposition of Guaiacol and Catechol
The theoretical aspects of the development of a chemical kinetic model for guaiacol and catechol pyrolysis are presented to describe the pyrolysis behaviors of the individual lignin-derived components. The possible pyrolysis pathways involving both unimolecular and bimolecular decomposition were investigated by the potential energy surfaces (PES) calculated at CBS-QB3 level. The high-pressure limiting rate constants of each elementary reaction step were evaluated based on the transition state theory (TST) to determine the dominant pyrolysis pathways. The kinetic analysis results predicted the most favorable catechol unimolecular decomposition pathways, where catechol isomerization to 2-hydroxycyclohexa-2,4-dien-1-one occurred via migration of the hydroxyl H atom, followed by decomposition into 1,3-cyclobutadiene, acetylene, and CO.
In the case of the bimolecular reaction of catechol, a hydrogen radical is coupled to the carbon atom in the benzene ring, leading to the formation of phenol and a hydroxyl radical through dehydroxylation. On the other hand, guaiacol is likely to form catechol and phenol via the O−CH3 homolysis and coupling of a hydrogen radical to the carbon atom with the methoxyl group, respectively.
Chapter 5: Theoretical Study on Elementary Reaction Steps in Thermal Decomposition Processes of Syringol-Type Monolignol Compounds
Chapter 5 presents a detailed theoretical chemical kinetic model (DCKM) to describe the vapor-phase pyrolytic behavior of several syringol-type monolignol compounds that are derived from the primary pyrolysis of lignin: 1-(4-hydroxy-3,5-dimethoxyphenyl)prop- 2-en-1-one (HDPP), sinapyl alcohol, 3-hydroxy-1-(4-hydroxy-3,5- dimethoxyphenyl)propan-1-one (HHDPP), 1-(4-hydroxy-3,5-dimethoxyphenyl)propane- 1,3-diol (HDPPD), and syringol. The possible pyrolytic pathways involving unimolecular decomposition, attack, and abstraction reactions were investigated by comparing the energy barriers calculated at the B3LYP/6-311++G(d,p) level. In the proposed pathways,
7
all syringol-type monolignols containing a side chain undergo its cleavage to form syringol through the formation of syringaldehyde or 4-vinylsyringol. Syringol is then converted into two products: (a) pyrogallol via the homolysis of the O-CH3 bond and hydrogenation; or (b) guaiacol via coupling reaction of a hydrogen radical with a carbon bearing methoxyl group in syrignol and the subsequent demethoxylation. The pyrolytic pathways of pyrogallol are classified into two processes: (a) the concerted dehydrogenation of the two hydroxyl H atoms and the unimolecular decomposition to produce acetylene (C2H2), ethynol (C2HOH), and CO; or (b) the displacement of an OH with H to produce catechol and resorcinol. The high-pressure limit rate constants for all the proposed elementary reaction steps were evaluated based on transition state theory.
Chapter 6: Theoretical Study on Elementary Reaction Steps in Thermal Decomposition Processes of Phenol-Type Monolignol Compounds
A detailed chemical kinetic model (DCKM) has been developed to theoretically predict the pyrolysis behavior of phenol-type monolignol compounds (1-(4-hydroxyphenyl)prop- 2-en-1-one, HPP; p-coumaryl alcohol, 3-hydroxy-1-(4-hydroxyphenyl)propan-1-one, HHPP; 1-(4-hydroxyphenyl)propane-1,3-diol, HPPD) released from the primary heterogeneous pyrolysis of lignin. The possible thermal decomposition pathways involving unimolecular decomposition, H-attack, and H-abstraction were investigated by comparing the activation energies calculated at the B3LYP/6-311++G(d,p) level of theory.
The results indicated that all phenol-type monolignol compounds convert to phenol by side-chain cleavage. p-Coumaryl alcohol decomposes into phenol via the formation of 4- vinylphenol, whereas HPP, HHPP, and HPPD decompose into phenol via the formation of 4-hydroxybenzaldehyde. The transition state theory rate constants for all the proposed elementary reaction channels were calculated in the high-pressure limit.
Chapter 7: Coupling Detailed Chemical Kinetic Models of Primary and Secondary Pyrolysis of Lignin
In this chapter, we construct the detailed chemical kinetic model (DCKM) predicting the lignin pyrolysis with both primary pyrolysis and secondary vapor-phase reactions by adding the DCKM of monolignol decompositions established in Chapter 3 – 6 into the semi-DCKM of primary pyrolysis described in Chapter 2. This model has improved the
8
prediction of the yields of inorganic gases (CO, CO2, H2O and H2), light organic compounds (CH4, C2H4, C3H6 and CH3OH), and aromatic compounds (C6H6 and C6H5OH).
Chapter 8: General Conclusions
The general conclusions in this thesis were summarized.
References:
[1] International Energy Agency, Home Page: https://www.iea.org/ (accessed October 3, 2017).
[2] Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass:
Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098.
[3] Boman, U. R.; Turnbull, J. H. Integrated biomass energy systems and emissions of carbon dioxide. Biomass and Bioenergy 1997, 13, 333–343.
[4] Kaltschmitt, M.; Reinhardt, G. A.; Stelzer, T. Life cycle analysis of biofuels under different environmental aspects. Biomass and Bioenergy 1997, 12, 121–134.
[5] Kuzuhara, Y. Biomass Nippon Strategy - Why “Biomass Nippon” now? Biomass Bioenergy 2005, 29, 331–335.
[6] Japan for Sustainability, Home Page:
https://www.japanfs.org/en/news/archives/news_id029799.html (accessed October 3, 2017).
[7] Sumitomo Corporation, Home page:
http://www.sumitomocorp.co.jp/english/news/detail/id=28944 (accessed October 3, 2017).
[8] McKendry, P. Energy production from biomass (part 2): conversion technologies.
Bioresour. Technol. 2012, 83, 47–54.
[9] Nussbaumer, T. Combustion and Co-combustion of Biomass: Fundamentals, Technologies, and Primary Measures for Emission Reduction. Energy and Fuels 2003, 17, 1510–1521.
[10] McKendry, P. Energy production from biomass (part 1): Overview of biomass.
Bioresour. Technol. 2002, 83, 37–46.
[11] Murugesan, A.; Umarani, C.; Subramanian, R.; Nedunchezhian, N. Bio-diesel as an alternative fuel for diesel engines-A review. Renew. Sustain. Energy Rev. 2009, 13, 653–
9
662.
[12] Naik, S. N.; Goud, V. V.; Rout, P. K.; Dalai, A. K. Production of first and second generation biofuels: A comprehensive review. Renew. Sustain. Energy Rev. 2010, 14, 578–597.
[13] Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism:
A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86.
[14] Lovell, A. B.; Glassman, K. B. I. Benzene oxidation perturbed by NO2 addition.
Symp. Combust. 1989, 22, 1063–1074.
[15] Ko, T.; Adusei, G. Y.; Fontjin, A. Kinetics of the O(3P) + C6H6 Reaction over a Wide Temperature Range. J. Phys. Chem. 1991, 95, 8745–8748.
[16] Nguyen, T. L.; Peeters, J.; Vereecken, L. Theoretical reinvestigation of the O(3P) + C6H 6 reaction: Quantum chemical and statistical rate calculations. J. Phys. Chem. A 2007, 111, 3836–3849.
[17] Seta, T.; Nakajima, M.; Miyoshi, A. High-temperature reactions of OH radicals with benzene and toluene. J. Phys. Chem. A 2006, 110, 5081–5090.
[18] Horn, C.; Roy, K.; Frank, P.; Just T. Shock-tube study on the high-temperature pyrolysis of phenol. Symp. Combust. 1998, 27, 321–328.
[19] Xu, Z. F.; Lin, M. C. Ab Initio Kinetics for the Unimolecular Reaction C6H5OH → CO + C5H6. J. Phys. Chem. A 2006, 110, 1672–1677.
10
Table 1: Biomass Process Options Classification.[8]
Conversion process Final products Advantages
Direct combustion Steam Most often-used conversion process Heat
Electric energy
Biochemical Bioethanol Useful method for moisture content biomass Biogas
Agrochemical Biodiesel Simple production process
gasification Steam High efficiency using combined-cycle gas turbine systems
Heat
Electric energy Fuel gas
pyrolysis Char-coal Conversion to various types of fuels Fuel gas
Bio-oil
11
Figure 1. Comparison of the rate constant of Rxn-3 given by Nguyen et al.[16] and Ko et al.[15]
Figure 2. Comparison of the rate constant of Rxn-7 given experimentally and theoretically by Seta et al.[17]
1.0 1.5 2.0 2.5 3.0 3.5
-33 -32 -31 -30 -29 -28 -27 -26
Tappe, M. et al. (1989) Nguyen, T.L. et al. (2007)
log k
1000/T (1/K)
0.4 0.6 0.8 1.0 1.2 1.4 1.6
-28.0 -27.5 -27.0 -26.5 -26.0 -25.5 -25.0
-24.5 Experiments by Seta, T. et al. (2006) Calculations by Seta, T. et al. (2006)
log k
1000/T (1/K)
12
Figure 3. Comparison of the rate constants of Rxn-7 given by Horn et al.[18] and Xu et al.[19]
0.4 0.5 0.6 0.7 0.8 0.9 1.0 1.1 1.2 1.3 -15
-10 -5 0 5 10
15 Horn, C. et al. (1998)
Xu, Z. F. et al. (2006)
log k
1000/T (1/K)
13
Chapter 2: Predicting Molecular Composition of Primary Product Derived From Fast Pyrolysis of Lignin with Semi-Detailed Kinetic Model
2.5. Introduction
After cellulose, lignin is the second most abundant component in biomass [1,2]. As a residue of the pulp and paper industry, huge amounts of lignin are available at low cost [2]. Although currently most lignin is burned to produce heat, thermal conversion processes, including pyrolysis, gasification and liquefaction, can be used to convert lignin or biomass into useful products, such as gas, char, liquid fuels and chemicals as well as heat [3–6]. Among these thermal conversion processes, pyrolysis is known as a common step to cause fragmentation of the lignin or biomass structure [7]. Pyrolysis is divided into two stages: (a) primary pyrolysis, where volatiles escape from biomass particles; and (b) secondary vapor-phase reactions, where the produced volatiles undergo further cracking, combine, or condense in the vapor phase.
A lot of studies have been carried out to identify the pyrolysis products generated from secondary vapor-phase reactions and to establish lumped kinetic models [7–10].
Caballero et al. [10] established a lumped kinetic model for the global secondary reaction of Kraft lignin by assuming a first-order reaction. However, lumped kinetic models established based on global product categories, such as char, tar, and gases, cannot describe the formation mechanisms for specific products at the molecular level.
A detailed chemical kinetic model (DCKM) of vapor-phase reactions based on elementary reactions [11–30] has been developed to overcome the limitations of the lumping approach and provide information on the pyrolysis behaviors of individual components.
Our group revealed that the DCKM was able to reproduce not only the yields of major products but also those of minor products such as aromatic hydrocarbons, which were obtained with a two-stage tubular reactor (TS-TR) connected to a gas chromatograph (GC) [11–13]. However, the DCKM has been limited to secondary vapor-phase reactions.
Thus, the molecular composition of the volatiles derived from fast pyrolysis has to be obtained as a boundary condition with the TS-TR setting a residence time of 0.1 s for vapor-phase reactions [11–13]. In order to enhance the versatility of the DCKM, it is necessary to expand the DCKM to include the primary pyrolysis stage.
For primary pyrolysis of lignin, lumped kinetic models of the decomposition reaction have been extensively reported in the literature [31–35]. However, these models did not allow prediction of the molecular composition of gases and tar components. Recently, Xiong et al. [36] performed computational fluid dynamics (CFD) simulations coupled
14
with distributed activation energy mode (DAEM) reaction kinetics for lab-scale bubbling bed biomass pyrolysis reactor, and revealed that the coupled CFD–DAEM system does not significantly increase computational overhead. A reliable kinetic model, which predict the yields and the molecular composition of gas and tar as accurately as possible, has been required with the development of highly efficient CFD methods [36–45] and the increase of computational resource. Faravelli et al. [46] explored a semi-detailed kinetic model, which characterizes lignin structures using three virtual compounds and involves approximately 100 species and 400 reactions, to predict the molecular compositions of products derived from primary pyrolysis. Recently, Hough et al. [47] added eight reactions into the kinetic models established by Faravelli et al. [46], and compared the integral yields of char, tar, and gases predicted by the model and observed for slow pyrolysis experiments by thermogravimetric analysis (TGA) [48]. However, the model predictions have not yet been compared with the molecular compositions of volatiles derived from fast pyrolysis experiments, such as TS-TR experiments. This comparison is also an important step to optimize fast pyrolysis process, which generates much amount of volatile products from biomass and produces “bio-fuel” [5].
The purpose of this study is to examine whether the semi-detailed kinetic model established by Hough et al. [47] could reproduce the molecular composition of the primary products generated from fast pyrolysis with TS-TR experiments. First, characterization of lignin structures was described using virtual compounds. Second, the heating rate for lignin samples in the primary pyrolysis zone of the TS-TR was estimated.
Finally, based on the estimated heating rate, the yields of char and volatiles were predicted using the semi-detailed kinetic model and compared with TS-TR experimental results [13] for the critical evaluation. This estimation would help to integrate the DCKM of both primary pyrolysis and secondary vapor-phase reactions.
2.6. Methods and Modeling
2.6.1. Virtual Compounds for Approximating Lignin Structures
A detailed primary pyrolysis analysis of three types of lignin (enzymatic hydrolysis lignin, EHL; organic extracted lignin, OEL; and Klason lignin, KL) with TS-TR was reported by Yang et al. [13,49], and thus these lignins were employed in this study. We followed the approach of Hough et al. [47] by choosing virtual compounds to approximate various possible lignin structures. Three reference components based on a β-O-4 skeleton [50]
(PLIG-C, PLIG-H, and PLIG-O, following the naming conventions of Hough et al. [47]) as shown in Figure 1 were selected to characterize the structures of EHL, OEL, and KL.
15
Figure 2 gives a synoptic view of the elemental compositions of the six lignins (PLIG-C, PLIG-H, PLIG-O, EHL, OEL, and KL). The elemental compositions of OEL and KL was inside the bounds of a triangle whose vertices were the three reference compounds. Thus, the equivalent compositions of OEL and KL can be described in terms of the three reference compounds using a linear relationship based on mass conservation. On the other hand, as the elemental composition of EHL was outside the bounds of the triangle, the mass fraction of PLIG-O had a negative value. In this case, the mass fraction of PLIG-O was set to zero and the values of the other two components (i.e. PLIG-C and PLIG-H) were normalized to express the equivalent composition of EHL. Table 1 lists the elemental compositions and the equivalent compositions of EHL, OEL, and KL.
2.6.2. Estimation of Heating Rate
No direct measurement of the sample heating rate has been made. Here, a numerical model for briefly estimating the heating rate and the results is presented. By assuming that the sample has a spherical shape, the temperature history (T) along the radial direction (r) in the lignin sample was estimated based on the following heat transfer equation:
𝜕𝑇
𝜕𝑡 = 𝛼
𝑟2
𝜕
𝜕𝑟(𝑟2𝜕𝑇
𝜕𝑟) (1)
The average temperature (Tav) in particle was expressed as:
𝑇𝑎𝑣 = 1
𝑅∫ 𝑇𝑑𝑟
𝑅 0
(2)
The following assumptions were made for this calculation.
1. The thermal diffusivity (α) and particle size (R) are 1.83×10−7 m2/s and 0.56 mm, which are calculated by solving the following equation:
α = 𝑘
𝜌𝐶𝑝 (3)
𝑚 = 𝜌4
3𝜋𝑟3 (4)
16
where the thermal conductivity (k), heat capacity (Cp), density (ρ), and mass (m) are 0.39 W/m/K [51], 1.60 J/g/K [52], 1.33 g/cm3 [53] and 1.00 mg [13], respectively.
2. The heat of reaction is negligibly small.
3. The initial temperature in the particle is 300 K.
4. When the particle is dropped into the bottom of the first isothermal zone (the primary pyrolysis zone) [13,49], the surface of the particle is exposed to the experimentally- defined pyrolysis temperature. Thus, the temperature at the wall (Ts) is fixed at 773–1223 K.
Using these four assumptions, Eqs. (1) and (2) were solved by the finite difference method, as described in the Appendix A.
Figure 3 shows the average temperature profile (Tav) as a function of time, as derived from Eq. (2). According to the temperature curves in Figure 3, the range of heating rate was estimated as 102–104 K/s.
2.6.3. Detailed Chemical Kinetic Model for Lignin Pyrolysis
The semi-detailed kinetic model proposed by Hough et al. [47] was used to predict the molecular composition of the primary products generated from fast pyrolysis with TS-TR experiments [13]. Details of the reactions and sources of kinetic parameters are available in Hough et al.’s article [47]. DETCHEMBATCH code [11,12] was used to run the proposed kinetic models. Numerical simulations were performed under various heating rates (102, 103, and 104 K/s). After heating up to the experimentally-designated temperature (773–
1223 K), the temperature was held isothermally until the char yield was constant.
Figure 4a–d shows the predicted thermal degradation behavior of EHL and product formation at 103 K/s heated up to 1223 K. Table 2 lists the chemical structures of the intermediates and products. Figure 4a and b revealed that above 700 K the intermediate components of LIGH and LIGC were formed immediately upon degradation of PLIG-C and PLIGH. As shown in Figure 4c and d, above 1000 K aromatic carbons (C10H2), VKETDM2, and H2O were generated as the main char, tar and gas products, respectively.
Here, volatile species that were heavier than benzene were defined as tar, otherwise done as gas. The formation of aromatic carbons in char well reproduced the product characteristics revealed by temperature-dependent nuclear magnetic (NMR) spectra [54,55]. Similarly, Figure. S3–S9 in Appendix A shows the predicted thermal degradation
17
behavior of three lignins (EHL, OEL and KL) and product formation under various temperature conditions.
Experimental data obtained with the TS-TR connected to GC [12,13] was used to compare with numerical predictions. The TS-TR consists of two zones: (a) first zone for the primary pyrolysis, and (b) secondary zone for the vapor-phase reactions of volatiles.
The sample was dropped into the bottom of the first zone, and then the generated volatiles were flown into the secondary zone with helium carrier gas. After passing through the secondary zone, the volatiles were directed into GC column and further identified by GC detector.
2.7. Results and Discussion
To critically evaluate the semi-detailed kinetic model for fast pyrolysis of lignin samples, the predicted yields were compared with experimental data. Figure 5 shows a comparison between the product yields (char, tar, CO, CO2 and H2O) at the terminal points in Figure S3–S7 and the experimental data [13] obtained using a TS-TR with a residence time of 0.1 s for vapor-phase reactions. The predicted chemical compositions of char and tar are described in Figure S10–S19 in the Appendix A. According to the data shown in Figure 5, the heating rate has a small effect on the yields. At all temperatures our numerical results for char and H2O yields agreed well with the experimental measurements (Figure 5a and e, respectively). However, the numerical simulation overestimated the tar yield and underestimated the CO yield at high temperature (Figure 5b and c). These differences probably result from a lack of the kinetic model of the secondary vapor-phase reaction, especially tar cracking reaction, which might not be negligible at high temperature. Thus, it is necessary to integrate the DCKM for the primary and secondary pyrolysis and to repeat the predictions for the yields of tar and CO. However, the kinetic parameters on the decomposition of monolignols being a major component of tar (Figure S10–S14 in the Appendix A) have not yet been investigated.
At all temperatures the simulation underestimated CO2 yield (Figure 5d). There are a number of possible reasons for this deviation. For instance, CO2 could be formed by the water-gas shift reaction (CO+H2O⇌CO2+H2) in the vapor phase which are not participate in the current model. Further, hemicellulose with xylan units that can act as CO2 sources [56] might exist as an impurity in the sample. Other possible reason is that the equivalent composition of EHL did not include a PLIG-O component, which include a carboxyl group leading to CO2 formation, as shown in Table 1. Figure 6 shows the change of the CO2 yield at 1223 K during the course of increasing the mass fraction of PLIG-O from
18
0.0 to 30.0 wt%. Because the CO2 yield increases with the increase of the mass fraction of PLIG-O, the addition of virtual compounds with the carboxyl group is expected to improve the prediction of the CO2 yield.
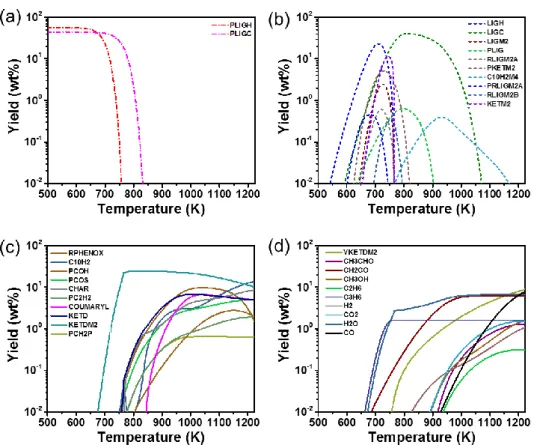
Figure 7a–c depicts the yields of methanol (CH3OH), ethane (C2H6), and propene (C3H6), respectively, which are minor products generated during EHL pyrolysis. As shown in Figure 7, the semi-detailed kinetic model proposed by Hough et al. [47] does not exactly reproduce the yields of these minor products. The kinetic database for the thermal decomposition processes of CH3OH, C2H6, and C3H6 has been already included in the DCKM of secondary vapor phase reactions established by our group [13], although which does not include that of monolignols. By using this DCKM, numerical simulations of secondary vapor-phase reactions of CH3OH, C2H6, and C3H6 were conducted at temperature of 773–1223 K and residence time of 0.1 s. The predicted yields at 103 K/s in Figure 7 were used as the initial species boundary. Figure 8a–c shows a comparison of the predicted yields of CH3OH, C2H6, and C3H6 between with and without the DCKM of the secondary vapor-phase reaction [13]. It is clear from Figure 8a–c that the numerical predictions were improved especially at high temperature by considering the secondary vapor-phase reactions.
Figure 9a–c shows a comparison of the predicted char, tar, and gas yields for the three lignin samples (EHL, OEL, and KL) with experimental data at 923 K. The experimental yields included in Figure 9 were obtained from TS-TR experiments with a residence time of 0.6 s for vapor-phase reactions [49]. Figure 9a and b clearly indicate that the model predictions reproduce the experimentally observed trend for char and tar yields, with the char yield increasing in order of EHL < OEL < KL, whereas the tar yields decrease.
However, a quantitative comparison could not be made because the effect of secondary vapor-phase reactions could not be neglected at a residence time of 0.6 s.
2.8. Conclusions
The semi-detailed kinetic model of lignin primary pyrolysis proposed by Hough et al.
[47] was used to predict the yield of char and volatile components (i.e., tar, CO, CO2, H2O, CH3OH, C2H6, and C3H6) obtained from EHL pyrolysis experiments in a TS-TR [13,49]. By solving the heat transfer equation, the heating rate of lignin particle in a TS- TR was estimated as 102–104 K/s. Numerical simulations were performed under various heating rates (102, 103, and 104 K/s) for temperatures of 773–1223 K. The numerical results for char and H2O yields were found to be in good agreement with the experimental results at all temperatures. However, the numerical simulation overestimated the tar yield
19
and underestimated the CO yield at high temperatures, probably owing to the absence of secondary vapor-phase cracking reactions in the model. The predicted CO2 yield was improved by adding PLIG-O with the carboxyl group into the reference compounds. The predicted yields of minor products (CH3OH, C2H6, and C3H6) showed good correspondence with the experimental values at high temperature by adding the DCKM of secondary vapor-phase reactions [13] into the semi-detailed kinetic model of lignin primary pyrolysis [47]. The char and tar yields derived from EHL, OEL, and KL at 923 K agreed qualitatively, but not quantitatively, with the experimental data. In the future, the characterization of lignin structure based on IR or NMR spectra as well as elemental composition should be examined for adding the virtual compounds with the carboxyl group. Additionally, further work to obtain the kinetic parameters of monolignol decompositions based on ab initio calculations and transition state theory should be implemented for establishing the DCKM for predicting lignin pyrolysis behavior with both primary pyrolysis and secondary vapor-phase reactions.
References:
[1] Rohella, R. S.; Sahoo, N.; Paul, S. C.; Choudhury, S.; Chakravortty, V. Thermal studies on isolated and purified lignin. Thermochim. Acta. 1996, 287, 131–8.
[2] Montané, D.; Torné-Fernández, V.; Fierro, V. Activated carbons from lignin: kinetic modeling of the pyrolysis of Kraft lignin activated with phosphoric acid. Chem. Eng. J.
2005, 106, 1–12.
[3] Azadi, P.; Inderwildi, O. R.; Farnood, R.; King, D. A. Liquid fuels, hydrogen and chemicals from lignin: a critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–23.
[4] Davis, K. M.; Rover, M.; Brown, R. C.; Bai, X.; Wen, Z.; Jarboe, L. R. Recovery and utilization of lignin monomers as part of the biorefinery approach. Energies 2016, 9, 1–
28.
[5] Mu, W.; Ben, H.; Ragauskas, A.; Deng, Y. Lignin pyrolysis components and upgradingtechnology review. Bioenergy Res. 2013, 6, 1183–1204.
[6] Pandey, M. P.; Kim, C. S. Lignin depolymerization and conversion: a review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41.
[7] Jegers, H. E.; Klein, M. T. Primary and secondary lignin pyrolysis reaction pathways.
Ind. Eng. Chem. Process Des. Dev. 1985, 24, 173–183.
[8] Hosoya, T.; Kawamoto, H.; Saka, S. Secondary reactions of lignin-derived primary tar components. J. Anal. Appl. Pyrol. 2008, 83, 78–87.
[9] Zhou, S.; Garcia-Perez, M.; Pecha, B.; McDonald, A. G.; Kersten, S. R. A.; Westerhof,
20
R. J. M. Secondary vapor phase reactions of lignin-derived oligomers obtained by fast pyrolysis of pine wood. Energy Fuels 2013, 27, 1428–1438.
[10] Caballero, J. A.; Font, R.; Marcilla, A. Kinetic study of the secondary thermal decomposition of Kraft lignin. J. Anal. Appl. Pyrol. 1996, 38, 131–152.
[11] Thimthong, N.; Appari, S.; Tanaka, R.; Iwanaga, K.; Kudo, S.; Hayashi, J.-I. et al.
Kinetic modeling of non-catalytic partial oxidation of nascent volatiles derived from fast pyrolysis of woody biomass with detailed chemistry. Fuel Process. Technol. 2015, 134, 159–167.
[12] Norinaga, K.; Yang, H.; Tanaka, R.; Appari, S.; Iwanaga, K,; Takashima, Y. et al. A mechanistic study on the reaction pathways leading to benzene and naphthalene in cellulose vapor phase cracking. Biomass Bioenergy 2014, 69, 144–154.
[13] Yang, H.; Appari, S.; Kudo S.; Hayashi, J.-I.; Norinaga K. Detailed chemical kinetic modeling of vapor-phase reactions of volatiles derived from fast pyrolysis of lignin. Ind.
Eng. Chem. Res. 2015, 54, 6855–6864.
[14] Shoji, T.; Norinaga, K.; Mašek, O.; Hayashi, J.-I. Numerical simulation of secondary gas phase reactions of coffee grounds with a detailed chemical kinetic model. Nihon Enerugi Gakkaishi/J Jpn. Inst. Energy 2010, 89, 955–961.
[15] Kohse-Höinghaus, K.; Oßwald, P.; Cool, T. A. Kasper, T., Hansen, N., Qi, F. et al.
Biofuel combustion chemistry: from ethanol to biodiesel. Angew. Chem. Int. Ed. 2010, 49, 3572–3597.
[16] Grana, R.; Frassoldati, A.; Faravelli, T.; Niemann, U.; Ranzi, E.; Seiser, R. et al. An experimental and kinetic modeling study of combustion of isomers of butanol. Combust.
Flame 2010, 157, 2137–2154.
[17] Harper, M. R.; Van Geem, K. M.; Pyl, S. P.; Marin, G. B.; Green, W. H.
Comprehensive reaction mechanism for n-butanol pyrolysis and combustion. Combust.
Flame 2011, 158, 16–41.
[18] Black, G.; Curran, H. J.; Pichon, S.; Simmie, J. M.; Zhukov, V. Bio-butanol:
combustion properties and detailed chemical kinetic model. Combust. Flame 2010, 157, 363–373.
[19] Labbe, N. J.; Seshadri, V.; Kasper, T.; Hansen, N.; Oßwald, P.; Westmoreland, P. R.
Flame chemistry of tetrahydropyran as a model heteroatomic biofuel. Proc. Combust. Inst.
2013, 34, 259–267.
[20] Lucassen, A.; Labbe, N.; Westmoreland, P. R.; Kohse-Höinghaus, K. Combustion chemistry and fuel-nitrogen conversion in a laminar premixed flame of morpholine as a model biofuel. Combust. Flame 2011, 158, 1647–1666.
[21] Li, W., Law, M. E.; Westmoreland, P. R.; Kasper, T.; Hansen, N.; Kohse-Höinghaus,
21
K. Multiple benzene-formation paths in a fuel-rich cyclohexane flame. Combust. Flame 2011, 158, 2077–2089.
[22] Kousoku, A.; Norinaga, K.; Miura, K. Extended detailed chemical kinetic model for benzene pyrolysis with new reaction pathways including oligomer formation. Ind. Eng.
Chem. Res. 2014, 53, 7956–7964.
[23] Norinaga, K.; Shoji, T.; Kudo, S.; Hayashi, J.-I. Detailed chemical kinetic modelling of vapour-phase cracking of multi-component molecular mixtures derived from the fast pyrolysis of cellulose. Fuel 2013, 103, 141–150.
[24] Norinaga, K.; Sakurai, Y.; Sato, R.; Hayashi, J.-I. Numerical simulation of thermal conversion of aromatic hydrocarbons in the presence of hydrogen and steam using a detailed chemical kinetic model. Chem. Eng. J. 2011, 178, 282–290.
[25] Norinaga, K.; Hayashi, J.-I. Numerical simulation of the partial oxidation of hot coke oven gas with a detailed chemical kinetic model. Energy Fuels 2010, 24, 165–172.
[26] Debiagi, P. E. A.; Gentile, G.; Pelucchi, M.; Frassoldati, A.; Cuoci, A.; Faravelli, T.
et al. Detailed kinetic mechanism of gas-phase reactions of volatiles released from biomass pyrolysis. Biomass Bioenergy 2016, 93, 60–71.
[27] Furutani, Y.; Kudo, S.; Hayashi, J.-I.; Norinaga, K. Theoretical study on reaction pathways leading to CO and CO2 in the pyrolysis of resorcinol. J. Phys. Chem. A 2017, 121, 631–637.
[28] Altarawneh, M.; Dlugogorski, B. Z.; Kennedy, E. M.; Mackie, J. C. Theoretical study of unimolecular decomposition of catechol. J. Phys. Chem. A. 2010, 114, 1060–1067.
[29] Liu H.; Chen J.; Wang F.; Wang Z.; Wang L. Theoretical study on the thermodynamic properties and stability of polybrominated diphenyl sulfide catena. Acta. Chim. Sin. 2010, 68, 540–550.
[30] Altarawneh, M.; Dlugogorski, B. Z.; Kennedy, E. M.; Mackie, J. C. Thermochemical properties and decomposition pathways of three isomeric semiquinone radicals. J. Phys.
Chem. A 2010, 114, 1098–1108.
[31] Klein, M. T.; Virk, P. S. Modeling of lignin thermolysis. Energy Fuels 2008, 22, 2175–2182.
[32] Nunn, T. R.; Howard, J. B.; Longwell, J. P.; Peters, W. A. Product compositions and kinetics in the rapid pyrolysis of sweet gum hardwood. Ind. Eng. Chem. Process. Des.
Dev. 1985, 24, 836–844.
[33] Petrocelli, F. P.; Klein, M. T. Model reaction pathways in Kraft lignin pyrolysis.
Macromolecules 1984, 17, 161–169.
[34] Caballero, J. A.; Font, R.; Marcilla, A. Study of the primary pyrolysis of Kraft lignin at high heating rates: yields and kinetics. J. Anal. Appl. Pyrol. 1996, 36, 159–178.
22
[35] Pasquali, C. E. L.; Herrera, H. Pyrolysis of lignin and IR analysis of residues.
Thermochim. Acta. 1997, 293, 39–46.
[36] Xiong, Q.; Zhang, J.; Xu, F.; Wiggins, G.; Daw, C. S. Coupling DAEM and CFD for simulating biomass fast pyrolysis in fluidized beds. J. Anal. Appl. Pyrol. 2016, 117, 176–
181.
[37] Xiong, Q.; Yang, Y.; Xu, F.; Pan, Y.; Zhang, J.; Hong, K. et al. Overview of computational fluid dynamics simulation of reactor-scale biomass pyrolysis. ACS Sustain.
Chem. Eng. 2017, 5, 2783–2798.
[38] Aramideh, S.; Xiong, Q.; Kong, S.-C.; Brown, R. C. Numerical simulation of biomass fast pyrolysis in an auger reactor. Fuel 2015, 156, 234–242.
[39] Mellin, P.; Kantarelis, E.; Yang, W. Computational fluid dynamics modeling of biomass fast pyrolysis in a fluidized bed reactor, using a comprehensive chemistry scheme. Fuel 2014, 117, 704–715.
[40] Trendewicz, A.; Braun, R.; Dutta, A.; Ziegler J. One dimensional steady-state circulating fluidized-bed reactor model for biomass fast pyrolysis. Fuel 2014, 133, 253–
62.
[41] Xiong, Q.; Aramideh, S.; Kong, S.-C. Modeling effects of operating conditions on biomass fast pyrolysis in bubbling fluidized bed reactors. Energy Fuels 2013, 27, 5948–
5956.
[42] Xiong, Q.; Aramideh, S.; Passalacqua, A.; Kong, S.-C. BIOTC: an open-source CFD code for simulating biomass fast pyrolysis. Comput Phys Commun 2014, 185, 1739–46.
[43] Xiong, Q.; Kong, S.-C. High-resolution particle-scale simulation of biomass pyrolysis. ACS Sustain. Chem. Eng. 2016, 4, 5456–5461.
[44] Xiong, Q.; Kong, S.-C. Modeling effects of interphase transport coefficients on biomass pyrolysis in fluidized beds. Powder Technol 2014, 262, 96–105.
[45] Xiong, Q.; Kong, S.-C.; Passalacqua, A. Development of a generalized numerical framework for simulating biomass fast pyrolysis in fluidized-bed reactors. Chem. Eng.
Sci. 2013, 99, 305–313.
[46] Faravelli, T.; Frassoldati, A.; Migliavacca, G.; Ranzi, E. Detailed kinetic modeling of the thermal degradation of lignins. Biomass Bioenergy 2010, 34, 290–301.
[47] Hough, B. R.; Schwartz, D. T.; Pfaendtner, J. Detailed kinetic modeling of lignin pyrolysis for process optimization. Ind. Eng. Chem. Res. 2016, 55, 9147–9153.
[48] Jakab, E.; Faix, O.; Till F. Thermal decomposition of milled wood lignins studied by thermogravimetry/mass spectrometry. J. Anal. Appl. Pyrol. 1997, 40–41, 171–186.
[49] Yang, H.; Appari, S.; Kudo, S.; Hayashi, J.-I.; Kumagai, S.; Norinaga, K. Chemical structures and primary pyrolysis characteristics of lignins obtained from different
23
preparation methods. Nihon Enerugi Gakkaishi/J Jpn. Inst. Energy 2014, 93, 986–994.
[50] Hage, R. E.; Brosse, N.; Chrusciel, L.; Sanchez, C.; Sannigrahi, P.; Ragauskas, A.
Characterization of milled wood lignin and ethanol organosolv lignin from miscanthus.
Polym. Degrad. Stab. 2009, 94, 1632–1638.
[51] Eitelberger, J.; Hofstetter, K. Prediction of transport properties of wood below the fiber saturation point – a multiscale homogenization approach and its experimental validation. Part I: Thermal conductivity. Compos. Sci. Technol. 2011, 71, 134–144.
[52] Hatakeyama, T.; Nakamura, K.; Hatakeyama, H. Studies on heat capacity of cellulose and lignin by differential scanning calorimetry. Polymer 1982, 23, 1801–1804.
[53] Youssefian, S.; Rahbar, N. Molecular origin of strength and stiffness in bamboo fibrils. Sci. Rep. 2015, 5, 11116.
[54] Lou, R.; Wu, S. B. Products properties from fast pyrolysis of enzymatic/mild acidolysis lignin. Appl. Energy 2011, 88, 316–322.
[55] Sharma, R. K.; Wooten, J. B.; Baliga, V. L.; Lin, X.; Chan, W. G.; Hajaligol, M. R.
Characterization of chars from pyrolysis of lignin. Fuel 2004, 83, 1469–1482.
[56] Shen, D. K.; Gu, S.; Bridgwater, A. V. Study on the pyrolytic behaviour of xylan- based hemicellulose using TG-FTIR and Py-GC-FTIR. J. Anal. Appl. Pyrol. 2010, 87, 199–206.
Table 1. Elemental compositions and equivalent compositions of EHL, OEL, and KL.
Elemental composition (wt%) Equivalent composition (wt%)
C H O PLIG-C PLIG-O PLIG-H
EHL 63.3 5.9 28.9 44.1 0.0 55.9
OEL 62.0 5.7 31.6 36.8 23.3 39.9
KL 62.9 5.3 31.4 55.3 41.3 3.4
24
Table 2. Main components of (a) intermediates and (b) products (following the naming conventions of Faravelli et al. [36]).
(a)
(b)
25
Figure 1. Virtual compounds used to characterize initial lignin structures [47].
Figure 2. Elemental composition of the six lignins.
26
Figure 3. Average temperature profiles (Tav [K]) as a function of time (t [s]) at Ts = 773 – 1223 K.
Figure 4. Yields of (a) reactants, (b) main intermediates, main products of (c) char and (d) volatile obtained by heating at 103 K/s to 1223 K. The chemical structures of intermediates and products are shown in Table 2.
0.0 2.0 4.0 6.0 8.0 10.0
400 600 800 1000 1200 1400 1600
1800 10
3 K/s 104 K/s
Average Temperature: Tav (K)
Time: t (s)
Ts=773 K Ts=923 K Ts=1023 K Ts=1123 K Ts=1223 K
102 K/s
27
Figure 5. Comparison of the predicted yields of (a) char, (b) tar, (c) CO, (d) CO2 and (e) H2O with experimental data [13].
28
1 2 3 4 5
0 2 4 6 8 10
30 wt%
20 wt%
10 wt%
Yield (wt%)
102 [K/s]
103 [K/s]
104 [K/s]
Experiment
0 wt%
Figure 6. The predicted change of the CO2 yield at 1223 K along with a fixed ratio of PLIG-C and PLIG-H and the mass fraction of PLIG-O increasing from 0.0 to 30.0 wt%.
The experimental data [13] was also shown to compare with the numerical predictions.
29
Figure 7. Comparison of the predicted yields of (a) CH3OH, (b) C2H6, and (c) C3H6
with experimental data [13].
30
Figure 8. Yields of (a) CH3OH, (b) C2H6, and (c) C3H6 predicted with and without DCKM of secondary vapor-phase reaction [13]. The experimental data [13] was also shown to compare with the numerical predictions.
31
Figure 9. Comparison of the predicted yields of (a) char, (b) tar, and (c) gas for EHL, OEL, and KL at a temperature of 923 K with experimental data [49].
32
Chapter 3: Theoretical Study on Reaction Pathways Leading to CO and CO2 in the Pyrolysis of Resorcinol
3.1. Introduction
Phenolic compounds are well-documented byproducts of the thermal degradation of biomass, particularly the degradation of lignin [1,2]. For example, dihydroxybenzene isomers present in cigarette smoke are biologically active and induce DNA damage, thereby causing serious health problems, such as lung cancer, heart disease, and oxidative stress [3−8].
As a model for the structural groups present in lignin, the pyrolysis of dihydroxybenzene isomers has been studied both experimentally and theoretically. For example, Thomas et al. [9] examined the formation of polycyclic aromatic hydrocarbons from the pyrolysis of 1,2-dihydroxybenzene (catechol). The formation of persistent semiquinone radicals (hydroxylsubstituted phenoxy radicals) during the pyrolysis of catechol and 1,4- dihydroxybenzene (hydroquinone) was observed by low-temperature matrix isolation electron paramagnetic resonance spectroscopy [10−12]. These semiquinone radicals contribute to the formation of notorious dibenzo-p-dioxins and dibenzofuran [13,14].
Recently, Yang et al. [15] performed pyrolysis experiments on catechol, hydroquinone, and 1,3-dihydroxybenzene (resorcinol) in a tubular reactor with a residence time of up to 3.6 s at 650−950 °C. Investigation of the product distribution showed that CO was the major product formed from catechol, resorcinol, and hydroquinone, with maximum yields of 27.3, 19.4, and 20.0 wt % at 950 °C over 0.3 s, respectively. Notably, the amount of CO2 (15.5 wt %) generated from resorcinol was significantly higher than that generated from hydroquinone (1.0 wt %) and catechol (0.8 wt %).
In addition to experimental studies, theoretical investigations using first-principles computational chemistry have been conducted to analyze the thermal decomposition pathways of dihydroxybenzene isomers. Alsoufi et al. [16] evaluated self-coupling reactions as the first step in the formation of dioxins from semiquinone radicals in terms of enthalpy and Gibbs free energy. In addition, unimolecular decomposition pathways on the potential energy surface (PES) of dihydroxybenzene isomers to yield CO have been investigated [11,17−19]. Indeed, CO formation via phenoxyl and semiquinone radicals to give cyclopentadienyl (cyc-C5H5) and hydroxycyclopentadienyl radicals (cyc-C5H4OH), respectively, has been reported [17−19]. In addition, Khachatryan et al. [11] suggested that the orthobenzoquinone generated from the O−H bond dissociation of ortho- semiquinone could form hexa-2,4-dienedial via ring-fission, resulting in the release of
33
CO. The fate of oxygen atoms in catechol and hydroquinone is essentially the formation of CO, while in resorcinol it is the formation of both CO2 and CO, as revealed by the experiments [15].
However, to date, the resorcinol decomposition pathways leading to CO2 formation upon pyrolysis are not yet to be theoretically identified. We herein describe investigations into the pyrolysis of resorcinol via CO2 and CO expulsion using first-principles computational chemistry. Initially, thermal decomposition pathways were proposed, focusing on the stages following the formation of the m-benzoquinone diradical. Finally, the PESs along the proposed reaction pathways were calculated using the CBS-QB3 procedure, and the high-pressure limiting rate constants were evaluated based on transition-state theory (TST).
3.2. Computational Method
All electronic structure calculations were performed using the Gaussian 09 computational chemistry software [20]. The PES of the proposed pathways was calculated using a series of high accuracy methods, including a complete basis set extrapolation, namely, the CBS- QB3 composite method [21]. In this CBS-QB3 method, geometries and frequencies were calculated at the B3LYP/6-311G(2d,d,p) level, which was important for the correct localization of transition structures. To approximate higher order contributions, the CBS- QB3 method uses two additional calculations, that is, MP4(SDQ)/6-31+G(d(f),p) and CCSD(T)/6-31+G†. The spin contamination and size consistency were also corrected. In addition, by combining the results of several electronic energy calculation steps, the CBS- QB3 offers greater accuracies within 1 kcal/mol [21].
Structures of transition state on the PES were confirmed by determination of only one imaginary frequency along the specific reaction coordinate through analysis of the vibration frequency. Intrinsic reaction coordinate (IRC) [22] calculations were performed to connect the related reactants and products.
Reaction rate constants at the high-pressure limit were calculated according to TST using the GPOP program [23]. 1D semiclassical tunneling corrections were included by assuming the asymmetric Eckart potential [24−26].
The variational TST (VTST) was used for the calculation of the rate constants for barrierless channels [23]. Energies along the reaction coordinates were evaluated by restricting the bond lengths and relaxing all of the other geometric parameters.
![Figure 8. Yields of (a) CH 3 OH, (b) C 2 H 6 , and (c) C 3 H 6 predicted with and without DCKM of secondary vapor-phase reaction [13]](https://thumb-ap.123doks.com/thumbv2/123deta/9915035.1917923/35.892.129.735.169.760/figure-yields-predicted-dckm-secondary-vapor-phase-reaction.webp)