©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
nature CHeMICaL BIOLOGY| ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemicalbiology 1
D
rug discovery is a challenging process that often spans 10–15 years1. Despite recent technological advances, the number of new molecular entities approved each year still falls below expectations2. Researchers in both industry and academia have tried to bypass the hurdles of drug discovery by repositioning clinical compounds for new therapeutic indications, a process commonly referred to as drug repurposing3.Patient diversity and drug resistance represent substantial obsta-cles in long-term pharmacological treatment of diseases such as cancer and infections4,5. Multicomponent therapeutics can be effec-tive alternaeffec-tives in these circumstances6, providing a new strategy for more personalized medicine. Approved drugs, which have been certified to be safe, effective and bioavailable and have well-defined targets and mechanisms of action, are an ideal starting point for the development of new multicomponent regimes and provide a fast track to new clinical applications.
These unique features have prompted efforts to catalog and col-lect all drugs approved for human or veterinary use7,8; however, access to these comprehensive libraries is limited. Moreover, a systematic pairwise combinatorial high-throughput screen (HTS) of all 14,814 molecules currently in the US National Institutes of Health Chemical Genomics Center (NCGC) pharmaceutical collec-tion8 would generate more than 100,000,000 data points, an effort beyond the current capacity of screening facilities.
In this study, we report a strategy to computationally derive a set of representative drugs that have been approved by the US Food
and Drug Administration (FDA). Our aim was to generate a collec-tion that optimally captures the biochemical diversity of all clinical compounds while fitting on a single 384-well screening plate. We annotated this collection with reported human peak plasma con-centrations to encourage HTS at pharmacologically relevant doses. Moreover, for all substances administered as prodrugs, we included the respective active form. We named this collection of clinical com-pounds the CeMM Library of Unique Drugs (CLOUD).
The nonredundant nature of our library allows systematic inves-tigation of all combinations of CLOUD compounds. In a combina-torial HTS assessing cancer cell viability, we uncovered a synergistic interaction between flutamide and PPC, approved drugs represent-ing antiandrogens and vitamin K antagonists in the CLOUD. We found that the synergy between these two drug classes was con-served in multiple prostate cancer cell lines. Concomitant adminis-tration of flutamide and PPC impaired the viability of prostate cells carrying AR mutations associated with resistance to therapy among patients. Our findings suggest that vitamin K antagonists could be repurposed in the clinic to address resistance to antiandrogens mediated by AR mutations.
RESULTS
The CLOUD is a representative library of approved drugs
To generate a representative library of clinical compounds, we retrieved all approved products from the Drugs@FDA Database (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm). This
1CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2Christian Doppler Laboratory for Chemical
Epigenetics and Anti infectives, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 3Christian Doppler
Laboratory for Molecular Stress Research in Peritoneal Dialysis, Department of Pediatrics and Adolescent Medicine, Medical University of Vienna, Vienna, Austria. 4Department of Surgery and Perioperative Sciences, Urology and Andrology, Umeå University Hospital, Umeå, Sweden. 5Department of Surgical
Sciences, Uppsala University, Uppsala, Sweden. 6Enamine Ltd., Kiev, Ukraine. 7Department of Laboratory Medicine, Medical University of Vienna, Vienna,
Austria. 8Max Planck Institute for Informatics, Saarland Informatics Campus, Saarbru¨cken, Germany. 9Present addresses: Cancer Research UK Cancer
Therapeutics Unit, The Institute of Cancer Research, London, UK (M.P.L.); Pharmacy Department, Innsbruck University Hospital, Innsbruck, Austria (P.M.); Chemical Computing Group, Inc., Cologne, Germany (F.K.); Gregor Mendel Institute, Vienna, Austria (G.D.); Faculté de Médecine, Université de Montpellier, and Institut de Recherche en Cancérologie de Montpellier, INSERM U1194, Institut régional du Cancer Montpellier, Montpellier, France (J.C.). *e-mail: skubicek@cemm.oeaw.ac.at
a combinatorial screen of the CLOud uncovers a
synergy targeting the androgen receptor
Marco p Licciardello
1,9, anna ringler
1, patrick Markt
1,9, Freya Klepsch
1,9, Charles-Hugues Lardeau
1,2,
sara sdelci
1, erika schirghuber
1,2, andré C Müller
1, Michael Caldera
1, anja Wagner
3, rebecca Herzog
3,
thomas penz
1, Michael schuster
1, Bernd Boidol
1,2, Gerhard dürnberger
1,9, Yasin Folkvaljon
4, pär stattin
4,5,
Vladimir Ivanov
6, Jacques Colinge
1,9, Christoph Bock
1,7,8, Klaus Kratochwill
3, Jörg Menche
1,
Keiryn L Bennett
1& stefan Kubicek
1,2*
Approved drugs are invaluable tools to study biochemical pathways, and further characterization of these compounds may lead to repurposing of single drugs or combinations. Here we describe a collection of 308 small molecules representing the diversity of structures and molecular targets of all FDA-approved chemical entities. The CeMM Library of Unique Drugs (CLOUD) cov-ers prodrugs and active forms at pharmacologically relevant concentrations and is ideally suited for combinatorial studies. We screened pairwise combinations of CLOUD drugs for impairment of cancer cell viability and discovered a synergistic interaction between flutamide and phenprocoumon (PPC). The combination of these drugs modulates the stability of the androgen recep-tor (AR) and resensitizes AR-mutant prostate cancer cells to flutamide. Mechanistically, we show that the AR is a substrate for g-carboxylation, a post-translational modification inhibited by PPC. Collectively, our data suggest that PPC could be
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
2 nature CHeMICaL BIOLOGY| ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemicalbiology
resource includes prescription and over-the-counter small molecules and therapeutic biologicals approved for human use, as well as drugs discontinued for reasons other than safety. First, we determined
and extracted 2,171 unique active pharmaceutical ingredients responsible for the biological effects of the 26,800 products retrieved from the database (Fig. 1a). To obtain a small molecule collection
26,800 FDA-approved products
2,171 active ingredients
1,415 unique small molecules
920 drugs with known
target Clustering
239 unique
drugs 34 drugs with
unknown target
35 active forms of prodrugs
CLOUD
STEAM
954 systemic small molecules STEAM
+ +
a
c
b STEAM CLOUD
Dihydrofolate reductase inhibitors
Histone deacetylase inhibitors
Dihydrofolate reductase inhibitors
Histone deacetylase inhibitors
10 20 30
Drugs (%)
<0.01
0.01–0.1 0.1–1 1–10
10–100 100–1,000
Peak plasma concentration (µM) 25
15
5
0 20 40 60 80 100
0 1 2
Violations of Lipinski's rule of five
Drugs (%)
STEAM CLOUD
d
3 4
−2 −1 0 1 2
DIPS score
DIPS score > median (58)
DIPS score < median (11)
0
>1,000 e
Clusters of STEAM drugs
N
N N
N N
N H
OH
H2N NH2
OO OH
O N
H OH
HN N
HN H2N
O O
O OH O
N N N
N N H
OH NH2
H2N
O
O OH O
N N
N H NH2
H2N
O O
O
N H H
N HO
O O
NH N
H NH
H O
O O
NH O O H
O S S N
N N
N N
N H
OH
H2N NH2
OO OH
O
NH N
H NH
H O
O O
NH O O H
O S
S N
H H
N HO
O O
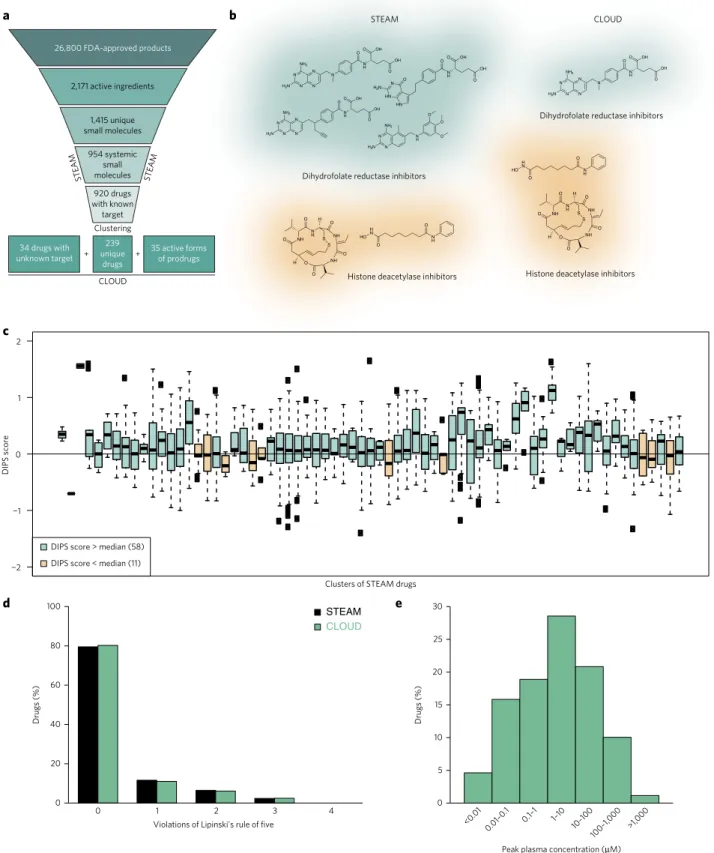
Figure 1 | The CLOUD. (a) Schematic representation of the filtering and clustering procedure leading to the 308 CLOUD drugs. (b) Examples of STEAM
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
nature CHeMICaL BIOLOGY| ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemicalbiology 3
suitable for HTS, we discarded all macromolecules, narrowing our set down to 1,928 small molecules. We also removed all salt frag-ments, which left 1,415 unique molecular entities. We then dis-carded all FDA-approved molecules that exert their biological effects through mechanisms other than protein–ligand interaction, are not used to treat diseases or are found only in topical products. This filtering scheme produced a collection of 954 systemically active small molecules, which we called STEAM (Supplementary Results,
Supplementary Data Set 1). To condense the STEAM into the CLOUD, we appended biological activities to all drugs with known molecular target and grouped them into classes (Supplementary Data Set 1). Each class is made up of drugs targeting the same protein family with the same mechanism of action. Compounds belonging to the same class are often structurally very similar owing to ‘me too’ drugs. While minor structural differences might change important pharmacological parameters in vivo, we reasoned that molecules that are structurally very similar and belong to the same activity class would behave redundantly in most screening assays. Therefore, we clustered all drugs within a specific group according to chemical structure and selected molecules at cluster centers. For example, the clustering algorithm selected methotrexate out of four structurally similar dihydrofolate reductase inhibitors (Fig. 1b). In contrast, vorinostat and romidepsin are distinct small molecule inhibitors of histone deacetylases. In this and similar cases we kept multiple compounds for the same target (Fig. 1b). Moreover, cer-tain drug classes cover a broader range of therapeutic activities: serotonin receptor agonists can be used to treat anxiety, migraine and disorders of gastrointestinal motility, depending on the spe-cific receptor subtype they engage. For such classes the clustering parameters were relaxed to allow for the selection of structurally related compounds covering a broader range of therapeutic indi-cations. Structural clustering of 176 classes resulted in 239 repre-sentative drugs optimized for chemical diversity and coverage of biological activities. To enable detection of compound activity in both biochemical and cellular assays, we searched the literature for prodrugs among these 239 molecules. We identified 35 prodrugs, for which the corresponding active forms were added to our screen-ing collection (Supplementary Table 1). Finally, the 34 STEAM drugs with unknown molecular target were included to generate the CLOUD collection of 308 small molecules (Supplementary Data Set 1 and Supplementary Table 1). The CLOUD covers more drug classes than do other commercially available libraries contain-ing FDA-approved drugs and exclusively provides active forms of prodrugs (Supplementary Fig. 1).
The CLOUD covers the biochemical space of approved drugs
To ensure that the CLOUD functionally preserved most of the bio-logical activities addressed by STEAM drugs, we analyzed gene expression profiles reported in the Connectivity Map (CMap)9 for compounds belonging to the same class. To eliminate batch effects from CMap data, we used drug-induced gene expression profile similarity (DIPS) scores10, which provide efficient data normaliza-tion. Pairwise DIPS scores within most STEAM clusters were higher than that of random drug pairs, indicating a similar influence of co-clustered drugs on cellular responses (Fig. 1c).
We then addressed the clustering procedure for potential biases in organisms, molecular targets or physicochemical proper-ties. The 920 STEAM drugs with known molecular targets engage mostly human proteins (Supplementary Fig. 2a). CLOUD drugs showed a similar distribution, with a minor reduction in bacte-ria-specific compounds owing to the high number of structurally similar antibiotics acting on dihydropteroate synthase, penicillin-binding proteins (PBP) and the ribosome. A molecular target clas-sification of STEAM drugs showed that the majority of approved small molecules engage G protein–coupled receptors (Supplementary Fig. 2b), as reported11. Again, CLOUD compounds showed a similar
pattern. Furthermore, orally bioavailable drugs usually adhere to ‘Lipinski’s rule of five’12, and approximately 80% of CLOUD and STEAM drugs did not violate this rule (Fig. 1d). Moreover, when we analyzed individually the physicochemical properties indi-cated in Lipinski’s rule of five and the numbers of rotatable bonds, we observed no significant differences in distribution between CLOUD and STEAM drugs (Supplementary Fig. 3). This con-firmed that the 239 CLOUD drugs with known molecular targets not only reflect the target distribution but also cover the chemical space of STEAM compounds.
Approved drugs have been extensively annotated with pharma-cological data, including human peak plasma concentrations. While biochemical and cellular HTS commonly apply compounds in the micromolar range, human peak plasma concentrations span several orders of magnitude (Fig. 1e). To foster clinically relevant discover-ies, we prepared stocks of CLOUD drugs enabling screens in the range of their respective plasma concentrations. Furthermore, we arranged all CLOUD compounds in a single 384-well plate with the aim of creating an easily accessible reference library for repurposing and annotating clinical drugs.
Flutamide and PPC interact synergistically
The nonredundant nature of the CLOUD enables effective combi-natorial screenings exploring new applications of approved drugs. In one such screen, we investigated the effect of pairwise com-binations of CLOUD compounds on the viability of KBM7 cells, a near-haploid human chronic myeloid leukemia (CML) cell line
PPC (µM) PPC (µM)
Flutamide (
µ
M)
Flutamide (
µ
M)
36.6 48.8 65.0 86.7
15.5 20.6 27.5 36.7
Fractional inhibition
0 1
27.4 20.6 15.4 11.6
11.6 6.5 8.7 4.9
36.6 48.8 65.0 86.7
15.5
20.6 27.5 36.7
–1 1
27.4 20.6 15.4 11.6
11.6 6.5 8.7 4.9
Deviation (Differential volume = 23.9) c
6
4
2
0
–2
–4
–6
–1 –0.5 0 0.5 1
Deviation
Log standard score
Flutamide PPC
0.0 0.5 1.0
–0.5
–1.0
Deviation
Synergy Antagonism
Flutamide PPC
a b
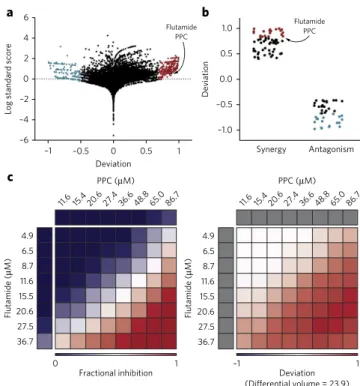
Figure 2 | A combinatorial HTS of the CLOUD uncovers the synergy
between flutamide and PPC. (a) Butterfly plot summarizing screen results.
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
4 nature CHeMICaL BIOLOGY| ADvAnCe onLine PubLiCAtion | www.nature.com/naturechemicalbiology
that enables the rapid downstream functional characterization of drug targets and mechanisms of action13,14 (Supplementary Table 2 and Supplementary Data Set 2). Using the ABL inhibitor dasat-inib as a positive control and measuring cellular ATP levels as a sur-rogate for cell viability, we tested 40,160 pairwise combinations and achieved robust screening results with an average Z′ factor of 0.4. In line with most drugs acting by mechanisms other than cytotoxic-ity, the majority of compounds did not affect the viability of KBM7 cells when used alone or in combination (Supplementary Fig. 4). We analyzed combinations of CLOUD compounds for potential synergy or antagonism according to the Bliss independence model15 (Fig. 2a). To corroborate the data, we also evaluated our results using the highest single agent (HSA) model16 and observed good correlation with the Bliss independence approach (Supplementary Fig. 5a). As most CLOUD drugs did not affect the viability of KBM7 cells even at the highest concentration tested, we could cal-culate Loewe scores17 for only 1,327 combinations. Predictions of synergy or antagonism according to the Chou–Talalay extension of Loewe additivity18 correlated with the Bliss independence model (Supplementary Fig. 5b). We further refined our results follow-ing recently published criteria based on a revision of the Saariselkä agreement19 (Supplementary Fig. 5c and Supplementary Data Set 3).
On the basis of the results from the Bliss independence analysis, we selected 254 synergistic and antagonistic hit combinations (Fig. 2a and Supplementary Table 2) for counter-screening and further inves-tigated the top 20 synergies and antagonisms in dose-response matrices (Fig. 2b and Supplementary Table 3). The synergistic interaction between flutamide, a nonsteroidal DHT-competitive AR antagonist approved for the treatment of prostate cancer, and PPC, a coumarin anticoagulant used for the treatment of thrombosis, stood out as the most compelling hit of the screen (Supplementary Fig. 6 and Fig. 2c).
KBM7 cells enable rapid generation of human gene knock-outs by insertional mutagenesis20,21. As flutamide targets the AR, we hypothesized that our synergistic interaction would rely on the presence of the nuclear receptor. We therefore addressed the effect of the drug combination on a KBM7 clone carrying a gene trap in the AR gene (ARGT KBM7). We first validated the loss of AR expression using RT-qPCR and western blotting (Supplementary Fig. 7a,b). Compared to wild-type KBM7 cells, ARGT KBM7 cells showed increased resistance to the combination of flutamide and PPC (Supplementary Fig. 7c). A closer inspection at the position of the gene-trap locus revealed an AR variant (AR45)22 downstream of the inserted cassette (Supplementary Fig. 7d). The expression of this isoform and residual transcripts of the full-length AR in ARGT KBM7 cells (Supplementary Fig. 7e) might explain the incomplete resistance to the combination.
AR and vitamin K antagonists synergize in prostate cancer
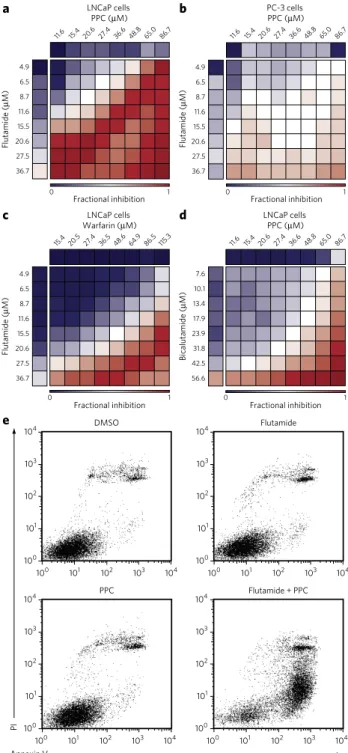
To evaluate the translational potential of the CLOUD and the syner-gistic interaction uncovered in our combinatorial screen, we assessed the effect of the combination of flutamide and PPC on various pros-tate cancer cell lines. We performed dose-response matrix measure-ments with LNCaP cells, an AR-dependent prostate cancer cell line carrying a T877A substitution in the AR23. LNCaP cells showed a marked sensitivity to the combination of flutamide and PPC (Fig. 3a and Supplementary Fig. 8a), whereas PC-3 cells, which express almost undetectable amounts of AR24, were only mildly affected at very high concentrations (Fig. 3b and Supplementary Fig. 8b). Flutamide and PPC also synergized to impair the viabil-ity of 22Rv1 cells, which harbor a duplication of exon 3 in the AR gene as well as a H874Y mutation in the AR C-terminal domain (Supplementary Fig. 9a). The two approved drugs synergized also in VCaP and LAPC4 cells, which express wild-type AR, albeit to a lower extent (Supplementary Fig. 9b,c). In contrast, DU145 cells, which express very low levels of AR, were not affected by the combi-nation, similarly to PC-3 cells (Supplementary Fig. 9d). To rule out
a contribution from basal AR signaling activation due to the pres-ence of endogenous ligands, we repeated the dose-response treat-ment in steroid-deprived conditions, confirming the sensitivity of LNCaP cells to the combination (Supplementary Fig. 10). Next, we
Flutamide (
µ
M)
Flutamide (
µ
M)
36.6 48.8 65.0 86.7
15.5 20.6 27.5 36.7
Fractional inhibition
0 1
27.4 20.6 15.4 11.6
11.6 6.5 8.7 4.9
36.6 48.8 65.0 86.7 27.4
20.6 15.4 11.6
15.5 20.6 27.5 36.7 11.6 6.5 8.7 4.9
Fractional inhibition
0 1
a LnCaP cells PPC (µM)
PC-3 cells PPC (µM)
b
c d
Flutamide (
µ
M)
Fractional inhibition
0 1
15.5 20.6 27.5 36.7 11.6 6.5 8.7 4.9
48.6 64.9 86.5 115.3 36.5
27.4 20.5 15.4
Fractional inhibition
0 1
36.6 48.8 65.0 86.7 27.4
20.6 15.4 11.6
bicalutamide (
µ
M)
23.9 31.8 42.5 56.6 17.9 10.1 13.4 7.6 LnCaP cells
Warfarin (µM)
LnCaP cells PPC (µM)
e
Annexin v
Pi
104
103
102
101
100
104
103
102
101
100 104
103
102
101
100 100 101 102 103 104
Flutamide DMSo
104
103
102
101
100
104
103
102
101
100
104
103
102
101
100 100 101 102 103 104
PPC Flutamide + PPC
Figure 3 | The combination of antiandrogens and vitamin K antagonists
induces apoptosis of LNCaP prostate cancer cells. (a,b) Dose-response
matrices for LnCaP (a) and PC-3 cells (b) treated with flutamide and PPC at the indicated concentrations for 3 d. (c,d) Dose-response matrices for LnCaP cells treated with flutamide and warfarin (c) or bicalutamide and PPC (d) at the indicated concentrations for 3 d. Data are average fractional inhibition of viability of 2 biological replicates (the corresponding deviations and differential volumes are illustrated in Supplementary
Figs. 8 and 11). (e) Propidium iodide–annexin v staining of LnCaP cells
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
nature CHeMICaL BIOLOGY| ADvAnCe onLine PubLiCAtion | www.nature.com/naturechemicalbiology 5
asked whether the synergistic compounds could be exchanged for other members of the drug classes they represent in the CLOUD. PPC was chosen as the representative vitamin K antagonist, a drug class that also includes warfarin. Of note, treatment of LNCaP cells with the combination of flutamide and warfarin resulted in a synergistic toxicity similar to that observed with PPC (Fig. 3c and
Supplementary Fig. 11a). Flutamide is the representative andro-gen receptor antagonist in the CLOUD; other STEAM drugs in this class include bicalutamide and nilutamide. When we tested these compounds in combination with PPC in LNCaP cells, we observed strong synergy with bicalutamide (Fig. 3d and Supplementary Fig. 11b). Enzalutamide and nilutamide also showed a synergistic interaction with PPC, albeit to a lower extent compared to flutamide (Supplementary Fig. 11c,d).
We then addressed whether PPC exerted its effect on the viabil-ity of LNCaP cells via inhibition of vitamin K epoxide reductase complex subunit 1 (VKORC1), the canonical mechanism of action
described for coumarin anticoagulants. Vitamin K functions as the cofactor of γ-glutamyl carboxylase (GGCX), which catalyzes
γ-carboxylation of glutamic acid residues in proteins involved in the coagulation cascade. VKORC1 converts oxidized vitamin K back to the reduced form sustaining the cellular pool of the cofactor25. The addition of vitamin K partially rescued the viability of LNCaP cells treated with flutamide and PPC in combination, with limited vitamin K solubility and uptake probably preventing complete antagonism (Supplementary Fig. 12a). Moreover, knockdown of GGCX increased the toxicity of flutamide in LNCaP cells but did not further decrease viability when flutamide was used in combina-tion with PPC (Supplementary Fig. 12b,c), supporting inhibition of protein γ-carboxylation as part of the mechanism underlying the synergistic interaction.
Propidium iodide–annexin V staining followed by flow cytometry analysis of LNCaP cells showed that only the concomitant adminis-tration of flutamide and PPC induced apoptosis in the AR-dependent
0 2 4 6
Relative mRnA level
AR KLK3 tMPRSS2 KLK2
8 10 20 40
130 kDa
100 kDa
35 kDa Wb: GAPDH
Wb: AR Flutamide PPC
– + +
+ +
–
– –
DAPi
AR Merge
DMSo
Flutamide
PPC
a b
c
β-tubulin
AR_FiRe
DAPi
AR_FiRe
DAPi
AR_FiRe
FiRe intensity
Low High
100 kDa
35 kDa Wb: GAPDH
Wb: AR
0 2 4 6 8 10 Hours
Flutamide + PPC
100 kDa
35 kDa
0 2 4 6 8 10
DMSo e
30
Flutamide PPC Flutamide + PPC
DMSo
130 kDa
100 kDa
35 kDa Wb: GAPDH
Wb: AR Flutamide PPC
– 30
– 5
30 5
15 10
15 10
vitamin K
– – + – +
f
Flutamide + PPC
Wb: AR Flutamide PPC
– + +
+ +
–
– –
130 kDa
100 kDa
35 kDa
+ +
– –
bortezomib d
DAPi
AR_FiRe
Wb: GAPDH
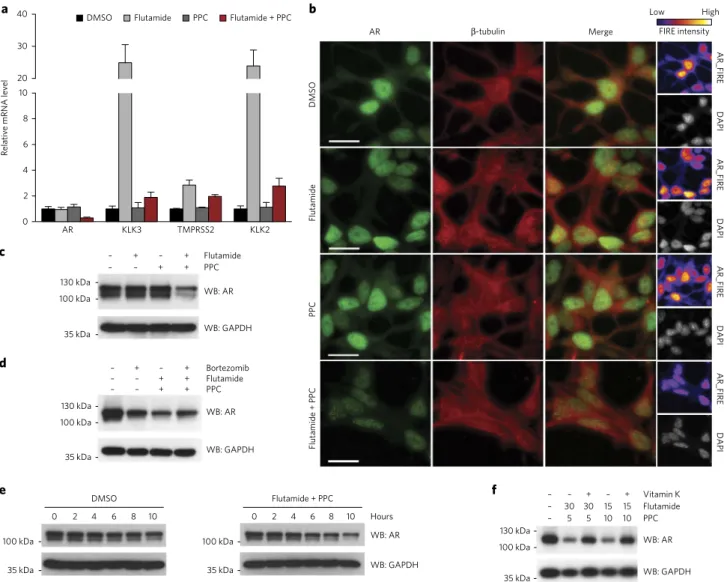
Figure 4 | The combination of flutamide and PPC induces AR degradation. (a) Rt-qPCR analysis of expression of AR and canonical targets of AR
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
6 nature CHeMICaL BIOLOGY| ADvAnCe onLine PubLiCAtion | www.nature.com/naturechemicalbiology
prostate cancer cells (Fig. 3e). Global gene expression profiling of LNCaP cells revealed that after 6 h, flutamide and PPC changed the expression of only a limited number of genes when used individu-ally. However, the administration of the drugs together affected 1,411 transcripts (Supplementary Fig. 13a). Gene set enrichment analy-sis26 identified apoptosis-associated terms to be significantly (false discovery rate q-value 0.002) upregulated when flutamide and PPC were used in combination (Supplementary Fig. 13b).
Overall, these data corroborate the concept behind the CLOUD and show that combinations of different antiandrogens and vitamin K antagonists synergistically lead to prostate cancer cell death.
Flutamide and PPC induce AR degradation
To further dissect the mechanism of action behind this synergistic interaction, we analyzed the effect of the combination on AR sig-naling. While flutamide normally acts as an AR antagonist, muta-tions in the AR ligand binding domain can switch flutamide activity to an agonistic one and have been described as a common resistance mechanism in prostate cancer27,28. RT-qPCR experiments confirmed that flutamide behaves as an AR agonist in LNCaP cells harbor-ing an AR T877A variant, as the drug increased the expression of AR signaling canonical targets such as KLK3, TMPRSS2 and KLK2 after 24 h (Fig. 4a). In contrast, PPC alone did not affect the expres-sion of these genes. Notably, the concomitant administration of flutamide and PPC restrained the expression of KLK3 and KLK2 (to levels similar to those observed with vehicle treatment) and decreased AR expression in LNCaP cells (Fig. 4a). As PPC alone did not interfere with the induction of AR signaling promoted by the potent synthetic agonist R1881 (Supplementary Fig. 14), we concluded that the vitamin K antagonist does not abolish binding of AR ligands to the receptor.
The decrease in AR mRNA upon treatment of LNCaP cells with the combination of flutamide and PPC could reflect a lower transcription rate or decreased mRNA stability. We assessed the turnover of AR mRNA in the presence of a transcription inhibi-tor over 10 h and found no difference between LNCaP cells treated with DMSO and those treated with the drug combination (Supplementary Fig. 15). In addition to transcripts, AR protein levels were reduced upon co-administration of flutamide and PPC, as assessed by immunofluorescence and western blotting (Fig. 4b,c
and Supplementary Fig. 16). Notably, AR mRNA and protein decreased after 8 h (Supplementary Fig. 17). Treatment of LNCaP cells with the proteasome inhibitor bortezomib reduced AR protein levels, as described29, and partly restored the levels of AR upon co-administration with flutamide and PPC (Fig. 4d). In addition, turn-over measurements revealed decreased AR half-life in LNCaP cells treated with the combination (Fig. 4e). Vitamin K partly rescued AR depletion in cells treated with flutamide and PPC and increased viability of LNCaP cells in a dose-dependent manner (Fig. 4f and
Supplementary Fig. 18). These results suggest that the concomitant administration of flutamide and PPC to LNCaP cells leads to AR degradation that occurs, at least in part, via the proteasome and by means of GGCX inhibition. As AR regulates its own expression30, transcriptional changes probably contribute to the overall decrease in protein abundance. Loss of AR has been described as sufficient to cause apoptosis of LNCaP cells31,32. In line with those findings, we were unable to derive viable LNCaP clones with stable AR knock-down or knockout. Short-term AR knockknock-down via small hairpin RNA (shRNA) in LNCaP cells showed induction of apoptosis after 120 h (Supplementary Fig. 19). The slower kinetics, as compared to the drug combination treatment, correlated with delayed loss of AR in the knockdown experiment.
The AR is post-translationally modified by
g
-carboxylation
PPC inhibits γ-carboxylation of proteins involved in blood coagu-lation. We hypothesized that this post-translational modification
could also occur on the AR and induce global conformational changes affecting protein stability and ligand binding.
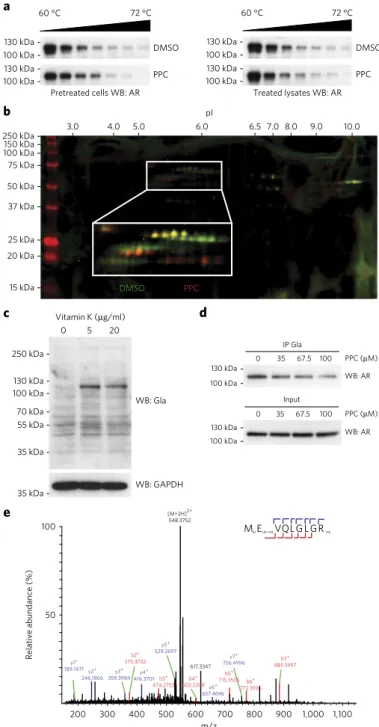
First, we evaluated the effect of PPC on the stability of the AR using a cellular thermal shift assay (CETSA)33, which confirmed that PPC affected the thermal stability of the AR
a 60 °C 72 °C
Pretreated cells Wb: AR DMSo
PPC 130 kDa
100 kDa 130 kDa 100 kDa
DMSo
treated lysates Wb: AR
60 °C 72 °C
PPC 130 kDa
100 kDa 130 kDa 100 kDa
b
15 kDa 20 kDa 25 kDa 37 kDa 50 kDa 75 kDa
100 kDa150 kDa
250 kDa
pi
3.0 4.0 5.0 6.0 6.5 7.0 8.0 9.0 10.0
DMSo PPC
c
Wb: Gla
vitamin K (µg/ml)
Wb: GAPDH 130 kDa
100 kDa
70 kDa 55 kDa
35 kDa 250 kDa
0 5 20
35 kDa
d
iP Gla
130 kDa 100 kDa
0 35 67.5 100 PPC (µM) input
Wb: AR 130 kDa
100 kDa
0 35 67.5 100 PPC (µM)
Wb: AR
e
Macecbx-mevQL G L G Rme
200 300 400 500 600 700 800 900 1,000 1,100
m/z 50
100
Relative abundance (%)
617.3347
y2+ 246.1866
y3+ 359.3984
y5+ 529.2697
b7+
885.3997
y4+ 416.3701
b5+
715.3505
b3+
474.2753 772.3926b6+
b2+
375.3732
y6+ 657.4696
y7+ 756.4996
y1+ 189.1471
b4+
602.2208
[M+2H]2+ 548.3752
Figure 5 | The AR is post-translationally modified by g-glutamyl
carboxylation. (a) CetSA followed by western blotting (Wb) of LnCaP
cells treated with DMSo or 35 μM PPC for 2 d (left) and LnCaP cell lysates treated with DMSo or 100 μM PPC for 30 min (right). (b) 2D difference gel electrophoresis (DiGe) of AR immunoprecipitated from LnCaP cells treated with DMSo (green) or PPC (red). (c) Western blotting for global
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
nature CHeMICaL BIOLOGY| ADvAnCe onLine PubLiCAtion | www.nature.com/naturechemicalbiology 7
(Fig. 5a and Supplementary Fig. 20). However, this shift in stability was observed only when we cultured LNCaP cells in the presence of PPC for 2 d before the experiment. In the absence of such pretreat-ment, the addition of PPC to the cell lysate did not stabilize the AR (Fig. 5a), suggesting there is not a direct engagement of the receptor by PPC. We then assessed whether PPC treatment could otherwise modify the AR and analyzed the migration pattern of the protein by means of 2D gel electrophoresis (Fig. 5b). We observed nonoverlap-ping spots when comparing AR immunoprecipitated from LNCaP cells pretreated with DMSO or PPC. Overall, AR from cells treated with PPC migrated at a higher isoelectric point, consistent with decreased negative charge. Both the CETSA and 2D gel electropho-resis results thus suggest post-translational modification changes on the AR induced by PPC.
Next, we assessed global γ-carboxylation to determine whether LNCaP cells are capable of γ-carboxylating protein substrates. Immunoblotting with an antibody recognizing γ-carboxylation in a sequence-independent context revealed several background bands over a wide range of molecular weights (Fig. 5c). However, addi-tion of vitamin K increased the abundance of only a few bands, the most prominent of which corresponded to the molecular weight of the AR (Fig. 5c). Notably, the same antibody immunoprecipitated the AR from LNCaP cell lysates, and PPC treatment reduced the amount of immunoprecipitated protein in a dose-dependent man-ner (Fig. 5d). Finally, to identify potential AR γ-carboxylation sites, we immunoprecipitated the receptor from LNCaP cells treated with vitamin K. We prepared the samples using procedures previously described for γ-carboxylated substrates34 and analyzed them by liq-uid chromatography–mass spectrometry. We detected a total of 19 AR-derived peptides and observed γ-carboxylation on E2 and E93 (Fig. 5e). These results show that the AR is γ-carboxylated and that this post-translational modification is inhibited by PPC.
DISCUSSION
In an attempt to physically obtain a complete set of all FDA-approved drugs for screening purposes, we realized that no commercially available compound collection covers all drug classes and that combinatorial screenings of even relatively small libraries would overload the infrastructure of most screening platforms. After an extensive literature search, we generated the CLOUD through cheminformatic selection of a set of FDA-approved drugs rep-resenting the entire target and chemical space of all clinical com-pounds. In contrast to existing collections, all CLOUD drugs fit on a single 384-well plate, are screened in the range of their human plasma concentration and include active forms of com-pounds clinically administered as prodrugs. At the CeMM chemical screening platform, we now use the CLOUD as a reference set in the development and optimization of all content and high-throughput assays.
The reductionist approach behind the CLOUD inevitably leads to the omission of additional targets and potential polypharmaco-logical features of some approved drugs. We have appended such additional information, based on a recent study35, to the descrip-tion of the library (Supplementary Data Set 1). The discovery of the synergistic interaction between flutamide and PPC in a combi-natorial HTS of the CLOUD, and the conservation of this synergy among other members of the drug classes these compounds repre-sent, validate the concept underlying our nonredundant collection of FDA-approved small molecules.
We identified the drug synergy in an AR-positive CML cell line and followed up in prostate cancer cells, as AR signaling has been shown to have a crucial role in the development of the disease36. Current prostate cancer pharmacological treatments aim at reduc-ing androgen levels or inhibitreduc-ing downstream signalreduc-ing with AR antagonists37,38. However, even though patients tend to respond to antiandrogens initially, they inevitably develop resistance39,40.
Drug combinations can circumvent or delay the development of resistance mechanisms, and the use of already approved drugs pro-vides different pharmacological and clinical benefits. We observed synergy between flutamide and PPC at clinically relevant con-centrations. While the maximum synergy occurs at concentra-tions higher than those observed in patients, LNCaP cell viability is clearly affected at the peak plasma concentrations of PPC and flutamide. Furthermore, periodic dosing and long-term treatment of patients is likely to result in higher cumulative exposure than that observed in 72 h experiments where single drug doses are administered to cancer cells in vitro. Our data therefore predict that patients receiving vitamin K antagonists are less likely to develop antiandrogen resistance and should therefore experience longer progression-free survival on these drugs. We retrospectively tested this hypothesis using records from the Prostate Cancer Database Sweden (PCBaSe)41. In brief, PCBaSe is a population-based cohort of more than 100,000 men with prostate cancer including data from the National Prostate Cancer Register, the Prescribed Drug Registry and the Cause of Death Registry. There was a nonsignifi-cant decrease in risk of prostate cancer death in 25 men treated with the combination for an extended time (Supplementary Fig. 21). Because the combination of these drugs in clinical practice is rare, larger cohorts are needed to provide final evidence for the clinical effects of the combination of antiandrogens with vitamin K antago-nists. Notably, similar attempts have tried to support evidence that warfarin use decreases the risk of prostate cancer mortality, but clinical results were inconclusive42. Ultimately, a thoroughly con-trolled prospective study together with rigorous patient stratifica-tion would be desirable to comprehensively address the clinical benefits of combining antiandrogens with vitamin K antagonists in the context of prostate cancer.
Mechanistically, we observed a reduction in AR protein stability and abundance upon treatment of LNCaP cells with the combina-tion of flutamide and PPC. The downregulacombina-tion of this important nuclear receptor is very likely to be the main apoptotic trigger in AR-dependent LNCaP cells. We also report that γ-carboxylation, which has previously been described only for a restricted group of proteins involved in the coagulation cascade, also occurs on at least two glutamic acid residues of the AR N-terminal domain. One of these AR modification sites was recently described by another group43. We show that γ-carboxylation alters the thermal stability and isoelectric point of the AR. Our findings support a model where PPC precludes
γ-carboxylation, and binding of flutamide to the uncarboxylated receptor results in a conformational change that induces protein degradation. Currently, we can only speculate on the conformational changes the AR undergoes before destabilization. The presence of
γ-carboxylation sites at the AR N terminus might be relevant for interactions with the ligand binding domain, which are known to be critical for AR activation. These interactions could be further influ-enced by mutations in the ligand binding domain. Indeed, flutamide and PPC show greater fitness impairment in LNCaP and 22Rv1 cells harboring mutations in the C terminus of the AR. Of note, resistance mutations in the ligand binding domain have been described to affect AR stability in response to different drugs44,45.
In summary, our results define a role for γ-carboxylation in AR signaling and suggest that vitamin K antagonists could be clinically repurposed for the formulation of more specific prostate cancer treatments.
Received 30 November 2015; accepted 2 March 2017; published online 22 May 2017
METHODS
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
8 nature CHeMICaL BIOLOGY| ADvAnCe onLine PubLiCAtion | www.nature.com/naturechemicalbiology
references
1. Paul, S.M. et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov.9, 203–214 (2010). 2. Scannell, J.W., Blanckley, A., Boldon, H. & Warrington, B. Diagnosing the
decline in pharmaceutical R&D eiciency. Nat. Rev. Drug Discov.11, 191–200 (2012).
3. Ashburn, T.T. & hor, K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov.3, 673–683 (2004). 4. Clavel, F. & Hance, A.J. HIV drug resistance. N. Engl. J. Med.350,
1023–1035 (2004).
5. Holohan, C., Van Schaeybroeck, S., Longley, D.B. & Johnston, P.G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer13, 714–726 (2013). 6. Bock, C. & Lengauer, T. Managing drug resistance in cancer: lessons from
HIV therapy. Nat. Rev. Cancer12, 494–501 (2012).
7. Chong, C.R., Chen, X., Shi, L., Liu, J.O. & Sullivan, D.J. Jr. A clinical drug library screen identiies astemizole as an antimalarial agent. Nat. Chem. Biol.
2, 415–416 (2006).
8. Huang, R. et al. he NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med.3, 80ps16 (2011).
9. Lamb, J. et al. he Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science313, 1929–1935 (2006). 10. Iskar, M. et al. Drug-induced regulation of target expression. PLoS Comput.
Biol.6, e1000925 (2010).
11. Overington, J.P., Al-Lazikani, B. & Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov.5, 993–996 (2006).
12. Lipinski, C.A., Lombardo, F., Dominy, B.W. & Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.46, 3–26 (2001). 13. Reiling, J.H. et al. A haploid genetic screen identiies the major facilitator
domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc. Natl. Acad. Sci. USA108, 11756–11765 (2011). 14. Winter, G.E. et al. he solute carrier SLC35F2 enables YM155-mediated DNA
damage toxicity. Nat. Chem. Biol.10, 768–773 (2014).
15. Bliss, C.I. he toxicity of poisons applied jointly. Ann. Appl. Biol.26, 585–615 (1939).
16. Berenbaum, M.C. What is synergy? Pharmacol. Rev.41, 93–141 (1989). 17. Loewe, S. Die quantitativen Probleme der Pharmakologie. Ergeb. Physiol.27,
48–187 (1928).
18. Chou, T.C. & Talalay, P. Quantitative analysis of dose-efect relationships: the combined efects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul.
22, 27–55 (1984).
19. Tang, J., Wennerberg, K. & Aittokallio, T. What is synergy? he Saariselkä agreement revisited. Front. Pharmacol.6, 181 (2015).
20. Bürckstümmer, T. et al. A reversible gene trap collection empowers haploid genetics in human cells. Nat. Methods10, 965–971 (2013).
21. Carette, J.E. et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science326, 1231–1235 (2009).
22. Dehm, S.M. & Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer18, R183–R196 (2011).
23. Veldscholte, J. et al. he androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which afects steroid binding characteristics and response to anti-androgens. J. Steroid Biochem. Mol. Biol.
41, 665–669 (1992).
24. Alimirah, F., Chen, J., Basrawala, Z., Xin, H. & Choubey, D. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: implications for the androgen receptor functions and regulation. FEBS Lett.
580, 2294–2300 (2006).
25. Suttie, J.W. Vitamin K-dependent carboxylase. Annu. Rev. Biochem.54, 459–477 (1985).
26. Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression proiles. Proc. Natl. Acad. Sci. USA102, 15545–15550 (2005).
27. Feldman, B.J. & Feldman, D. he development of androgen-independent prostate cancer. Nat. Rev. Cancer1, 34–45 (2001).
28. Gaddipati, J.P. et al. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res.54, 2861–2864 (1994).
29. Bhattacharyya, R.S., Krishnan, A.V., Swami, S. & Feldman, D. Fulvestrant (ICI 182,780) down-regulates androgen receptor expression and diminishes androgenic responses in LNCaP human prostate cancer cells. Mol. Cancer her.5, 1539–1549 (2006).
30. Grad, J.M., Lyons, L.S., Robins, D.M. & Burnstein, K.L. he androgen receptor (AR) amino-terminus imposes androgen-speciic regulation of AR gene expression via an exonic enhancer. Endocrinology142, 1107–1116 (2001).
31. Eder, I.E. et al. Inhibition of LncaP prostate cancer cells by means of androgen receptor antisense oligonucleotides. Cancer Gene her.7, 997–1007 (2000).
32. Liao, X., Tang, S., hrasher, J.B., Griebling, T.L. & Li, B. Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol. Cancer her.4, 505–515 (2005).
33. Martinez Molina, D. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shit assay. Science341, 84–87 (2013). 34. Hallgren, K.W., Zhang, D., Kinter, M., Willard, B. & Berkner, K.L.
Methylation of γ-carboxylated Glu (Gla) allows detection by liquid chromatography-mass spectrometry and the identiication of Gla residues in the γ-glutamyl carboxylase. J. Proteome Res.12, 2365–2374 (2013). 35. Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev.
Drug Discov.16, 19–34 (2017).
36. Taplin, M.-E. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat. Clin. Pract. Oncol.4, 236–244 (2007).
37. de Bono, J.S. et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med.364, 1995–2005 (2011).
38. Tran, C. et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science324, 787–790 (2009). 39. Chen, C.D. et al. Molecular determinants of resistance to antiandrogen
therapy. Nat. Med.10, 33–39 (2004).
40. Balbas, M.D. et al. Overcoming mutation-based resistance to anti-androgens with rational drug design. eLife2, e00499 (2013).
41. Van Hemelrijck, M. et al. Cohort Proile: the National Prostate Cancer Register of Sweden and Prostate Cancer data Base Sweden 2.0. Int. J. Epidemiol.42, 956–967 (2013).
42. Tagalakis, V., Tamim, H., Blostein, M., Hanley, J.A. & Kahn, S.R. Risk of prostate cancer death in long-term users of warfarin: a population-based case-control study. Cancer Causes Control24, 1079–1085 (2013).
43. Tew, B.Y. et al. Vitamin K epoxide reductase regulation of androgen receptor activity. Oncotarget8, 13818–13831 (2017).
44. Itsumi, M. et al. PMA induces androgen receptor downregulation and cellular apoptosis in prostate cancer cells. J. Mol. Endocrinol.53, 31–41 (2014).
45. Yu, Z. et al. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin. Cancer Res.20, 4075–4085 (2014).
acknowledgments
We thank M. Iskar (EMBL) and P. Bork (EMBL) for providing us with DIPS scores, R. Schüle (Albert-Ludwigs-University Freiburg, Germany) for LAPC4 cells, and G. Winter (CeMM) and G. Superti-Furga (CeMM) for thoughtful discussions and initializing synergy screenings at CeMM. S.K. acknowledges support by a Marie Curie Career Integration Grant, the Austrian Federal Ministry of Science, Research and Economy and the National Foundation for Research, Technology, and Development and the Austrian Science Fund (FWF): F4701-B20.
author contributions
P.M., F.K. and S.K. designed and assembled the CLOUD. M.P.L., F.K., C.-H.L. and S.K. designed and performed the screen of the CLOUD. M.P.L., F.K., M.C. and J.M. analyzed the data from the screen. M.P.L. and A.R. designed and performed viability, RT-qPCR, western blotting, immunoprecipitation and immunofluorescence experiments. S.S. performed immunofluorescence experiments. A.R. and E.S. designed and performed gene expression and immunoprecipitation experiments. A.R. and B.B. designed and per-formed knockdown experiments. A.C.M. and K.L.B. designed and perper-formed proteomics experiments. A.W., R.H. and K.K. performed and analyzed 2D gel electrophoresis experiments. T.P., M.S. and C.B. performed and analyzed RNA-seq experiments. G.D. and J.C. performed DIPS score analyses. Y.F. and P.S. analyzed patient data in PCBaSe. V.I. synthesized, provided and quality controlled chemicals. M.P.L. and S.K. wrote the manuscript.
Competing financial interests
The authors declare competing financial interests: details accompany the online version
of the paper.
additional information
Any supplementary information, chemical compound information and source data are
available in the online version of the paper. Reprints and permissions information is
available online at http://www.nature.com/reprints/index.html. Publisher’s note: Springer
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
nature CHeMICaL BIOLOGY doi:10.1038/nchembio.2382
penicillin–streptomycin (Sigma-Aldrich). ARGT KBM7 cells were obtained from Haplogen. LNCaP (ATCC, CRL-1740), PC-3 (ATCC, CRL-1435), 22Rv1 (ATCC, CRL-2505) and DU145 (ATCC, HTB-81) cells were grown in RPMI-1640 supplemented with 10% FBS and 1% penicillin–streptomycin. VCaP (ATCC, CRL-2876) cells were grown in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. LAPC4 cells (kind gift from R.S.) were grown in Iscove’s modified Dulbecco’s medium supplemented with 10% FBS and 1% penicillin–streptomycin. For steroid-deprived experiments, LNCaP cells were cultured in RPMI-1640 supplemented with 10% charcoal stripped FBS (Thermo Fisher Scientific) and 1% penicillin–streptomycin.
Generation of the STEAM. The complete list of all FDA-approved products
was downloaded from the Drugs@FDA database (accessed 18 January 2011), and the names of all active ingredients were extracted. Information on bio-logical activity, Chemical Abstracts Service registry numbers, canonical smiles, and molecular weights was retrieved mainly from the DrugBank (https://www. drugbank.ca/) or the Therapeutic Target Database (http://bidd.nus.edu.sg/ group/cjttd/). Other sources included Clarke’s Analysis of Drugs and Poisons, the KEGG drug database, the Handbook of Clinical DrugData, and Martindale. Active ingredients were filtered as described in the text to obtain 954 systemi-cally active small molecules.
Generation of the CLOUD. Structural clustering of the 920 STEAM drugs
with known targets was performed using a Pipeline Pilot protocol (Accelrys). Structures were represented as extended connectivity fingerprints (ECFP), and the Tanimoto coefficient was applied as a measurement of structural distance. For cluster formation, the Tanimoto dissimilarity was set to 0.85. If this thresh-old could not cover most of the therapeutic activities within a drug class, the threshold was lowered in a stepwise procedure. The script was used to identify the cluster centers within each of the 176 drug classes. All cluster centers were kept, providing a final set of 239 structurally unique CLOUD drugs. To these, we added 34 STEAM drugs with unknown targets and the active forms of 35 prodrugs. Compounds were purchased mainly from Enamine Ltd, Toronto Research Chemical and Sigma-Aldrich. Controlled substances and unstable or unavailable compounds are indicated in Supplementary Data Set 1 and
Supplementary Table 1.
Physicochemical property calculations. Molecular weight, logP, the number
of rotatable bonds, hydrogen bond acceptors or donors and violations of the Lipinski’s rule of five were calculated using the chemistry components of the software Pipeline Pilot (Accelrys).
KBM7 cell viability screen. CLOUD drugs and combinations thereof were
trans-ferred on 384-well plates using an acoustic liquid handler (Echo, Labcyte) and 5,000 cells per well were dispensed on top of the drugs using a dispenser (Thermo Fisher Scientific) for a total of 50 μl/well. Viability was measured after 72 h using CellTiter-Glo (Promega) in a multilabel plate reader (EnVision, PekinElmer). Signal was then normalized to negative (DMSO) and positive (10 μM dasatinib) controls and set between 0 and 100. Noisy compounds, defined according to median absolute deviation (MAD), were excluded from the analy-sis together with their corresponding combinations. Drug combinations were analyzed according to the Bliss independence model15. Briefly, the effect of the combination of drug A and drug B can be predicted to be C = A + B − A × B, where A and B are the effects of the single drugs expressed as fractional inhibi-tion of viability between 0 and 1. A deviainhibi-tion of the experimental value from the Bliss prediction was calculated. Positive deviations denote synergies, while negative deviations indicate antagonisms. Top hits were selected setting thresh-olds for deviation (>0.7 for synergies and less than −0.5 for antagonisms) and
Z score (>1). Furthermore, we also calculated scores for drug combinations according to the highest single agent (HSA)16, the Loewe additivity model17,18 and criteria based on a revisited Saariselkä agreement19 as already described.
In all, 254 hits were selected and tested again in a counter-screen. The top 20 validated synergies and antagonisms were further validated in 4 ×
Differential volumes >1 were taken as synergies, and differential volumes less than −1 were considered antagonisms.
Reverse transcription quantitative polymerase chain reaction. RT-qPCR was
performed as described46. Primers are listed in Supplementary Table 4.
Western blotting. Cells were lysed in RIPA buffer supplemented with a
cock-tail of protease inhibitors (Roche). Lysates were resolved by SDS–PAGE. AR-specific (5153) and GAPDH-specific (5174) antibodies were purchased from Cell Signaling Technology. αTUB-specific (ab7291) and GGCX-specific (ab197982) antibodies were purchased from Abcam. Anti-γ-carboxyglutamyl antibody (3570) was purchased from Sekisui Diagnostics.
Immunofluorescence. Cells were grown on coverslips, fixed with methanol
at −20 °C for 24 h, washed once with PBS, permeabilized in solution-B (3% BSA in PBS, 0.1% Triton X-100, 0.02% azide) for 30 min and incubated with pri-mary antibodies in solution-B for 30 min at room temperature. βTUB-specific antibody (T4026, dilution 1:1,000) was purchased from Sigma-Aldrich, and AR-specific antibody (5153, dilution 1:500) was purchased from Cell Signaling Technologies. Primary antibodies were detected using Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG (Thermo Fisher Scientific) diluted in solution-B. DNA was stained with DAPI (Thermo Fisher Scientific, 0.01 mg/ml). Images were taken using a Leica DMI6000 B inverted confocal system and a 63× 1.30 ACS Apo lens, and edited using Leica LAS AF software (Leica Microsystems) and Fiji (ImageJ).
Apoptosis assay. In all, 50,000 cells/well were seeded in 6-well plates and
treated with DMSO, 15 μM flutamide, 35 μM PPC or flutamide and PPC in combination after 24 h. After 3 d, cells were labeled with Alexa Fluor 647 annexin V–specific antibody (BioLegend) and propidium iodide and analyzed using flow cytometry (BD FACSCalibur, BD Biosciences). Caspase3/7 activity was measure by Caspase-Glo 3/7 Assay (Promega) according to the manufac-turer’s protocol.
Cellular thermal shift assay. LNCaP cells were treated with DMSO or 35 μM
PPC for 2 d and then harvested maintaining a constant concentration of PPC in all subsequent handling buffers. Otherwise, lysates were directly treated with the drug as described33.
Immunoprecipitation. Cells were lysed in immunoprecipitation (IP) buffer
(50 mM Tris, pH 8, 150 mM NaCl, 1% IGEPAL) supplemented with a cock-tail of protease inhibitors (Roche). Immunoprecipitations were performed at 4 °C and using Dynabeads Protein G (Thermo Fisher Scientific). Lysates were precleared with beads for 30 min, incubated with antibodies overnight (1 μg antibody was used for 50 μg protein) and immunoprecipitated for 2 h. After washing, immunoprecipitated proteins were eluted either by boiling in Laemmli buffer or with 50 mM glycine, pH 2.8.
Gene expression profiling. LNCaP cells were treated with DMSO, 15 μM
flutamide, 35 μM PPC or the combination for 6 h. Cells were lysed and RNA isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The amount of total RNA was quantified using a Qubit 2.0 Fluorometric Quantitation system (Thermo Fisher Scientific), and the RNA integrity number (RIN) was determined using an Experion Automated Electrophoresis System (Bio-Rad).
©
2017 Nature America, Inc., part of Springer Nature. All rights reserved.
nature CHeMICaL BIOLOGY doi:10.1038/nchembio.2382
2D gel electrophoresis. IP lysates eluted with glycine buffer were used for 2D
difference gel electrophoresis (DIGE). Buffer was exchanged to DIGE sample buffer (30 mM Tris, pH 8.5, 7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopro-pyl) dimethylammonio]-1-propanesulfonate (CHAPS), 1 mM EDTA) using ultrafiltration columns (Millipore), and pH was adjusted to 8.5 as previously described49. Proteins were labeled with 200 pmol cyanine dyes (Refraction-2D, NH DyeAGNOSTICS) according to the manufacturer’s protocol. Rehydration and isoelectric focusing on IPG strips (ReadyStrip IPG Strips, 11 cm, pH 3–10, nonlinear (Bio-Rad)) was followed by SDS–PAGE on gradient gels (Criterion, Any kDa, Bio-Rad) and detection with a Typhoon fluorescence scanner (GE Healthcare) at the recommended excitation and emission wavelengths for G-Dye200 and G-Dye300. Images were analyzed using Delta2D 4.5 software (Decodon).
Knockdown experiments. Lentiviral vectors harboring shRNA sequences in a
pLKO.1 backbone were obtained from Sigma-Aldrich.
AR_shRNA1: CCGGCACCAATGTCAACTCCAGGATCTCGAGATCCT GGAGTTGACATTGGTGTTTTT
AR_shRNA2: CGGCACCAATGTCAACTCCAGGATCTCGAGATCCTG GAGTTGACATTGGTGTTTTTG
Liquid chromatography–mass spectrometry. Tryptically digested AR was
analyzed on a nano-high-performance liquid chromatography system (Agilent Technologies) coupled to a linear trap quadrupole Orbitrap Velos mass spec-trometer (Thermo Fisher Scientific) essentially as described50.
Data availability. RNA-seq data are available in Gene Expression Omnibus
(GEO) under accession number GSE94783.
46. Licciardello, M.P. et al. NOTCH1 activation in breast cancer confers sensitivity to inhibition of SUMOylation. Oncogene29, 319 (2014). 47. Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence
of insertions, deletions and gene fusions. Genome Biol.14, R36 (2013). 48. Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments.
Bioinformatics28, 2184–2185 (2012).
49. Rudashevskaya, E.L. et al. A method to resolve the composition of heterogeneous ainity-puriied protein complexes assembled around a common protein by chemical cross-linking, gel electrophoresis and mass spectrometry. Nat. Protoc.8, 75–97 (2013).